





Abstract
Congenital diaphragmatic hernia (CDH) is a rare congenital anomaly characterised by a diaphragmatic defect, persistent pulmonary hypertension (PH) and lung hypoplasia. The relative contribution of these three elements can vary considerably in individual patients. Most affected children suffer primarily from the associated PH, for which the therapeutic modalities are limited and frequently not evidence based. The vascular defects associated with PH, which is characterised by increased muscularisation of arterioles and capillaries, start to develop early in gestation. Pulmonary vascular development is integrated with the development of the airway epithelium. Although our knowledge is still incomplete, the processes involved in the growth and expansion of the vasculature are beginning to be unravelled. It is clear that early disturbances of this process lead to major pulmonary growth abnormalities, resulting in serious clinical challenges and in many cases death in the newborn. Here we provide an overview of the current molecular pathways involved in pulmonary vascular development. Moreover, we describe the abnormalities associated with CDH and the potential therapeutic approaches for this severe abnormality.
Abstract
Congenital pulmonary vascular diseases originate early during development, leading to clinical challenges http://ow.ly/EFno30hBhbv
Normal pulmonary vascular development
Human lung development can be divided into different stages based on histology, starting with the embryonic stage at 4 weeks of gestation, followed by the pseudoglandular stage in which branching of the lung buds continues. During the canalicular stage, starting at ∼16 weeks of gestation, the terminal bronchioli are formed. From 24 weeks of gestation until term, the saccular stage, airspaces widen and alveoli are formed. During the alveolar stage, which persists into the postnatal period up to 3 years of age, maturation of the airways occurs [1]. Concomitant with the expansion of the airways is the adaptation of the microvasculature to optimise the exchange of oxygen and carbon dioxide between the blood and the airways. We and others have shown that very early in lung development pulmonary vessels are present and connected to the systemic circulation. The vasculature develops in close relation with the airways and is a rate-limiting factor in branching morphogenesis [2–4]. This implies that the pulmonary vasculature plays an important role in lung development. The formation of new blood vessels primarily occurs through sprouting of new capillaries from pre-existing vessels at the growing tips of the branching epithelium, a process referred to as distal angiogenesis [4]. The newly formed endothelial tubes need to be stabilised by perivascular cells, pericytes, to become functional capillaries in a platelet derived growth factor-β dependent manner [5, 6]. Little is known about the pericyte population during lung development, but pericytes have been shown to originate from Wnt2+/Gli1+/Isl1+ multipotent cardiopulmonary mesoderm progenitors [7].
Pulmonary vascular resistance is high during gestation and decreases rapidly after birth under the influence of breathing movements, gas exchange and the release of vasoactive factors of the endothelin (ET), nitric oxide (NO) and prostacyclin (PGI2) pathways [8, 9] (figure 1). The ET pathway is activated by three ligands: ET-1, ET-2 and ET-3, of which ET-1 is the most common isoform [10, 11]. The ET-1 precursor protein, prepro-ET-1, is cleaved by furin into big-ET-1, which is subsequently processed to its active form by the endothelin-converting enzyme-1 and binds and activates two different G-protein coupled receptors, ETA and ETB, with equal affinity. The ETA receptor is located at the cell surface of vascular smooth muscle cells and induces vasoconstriction and cell proliferation by activating phospholipase C, whereas the ETB receptor is located mainly in the cell membrane of the endothelium and induces vasodilation by regulating the release of NO and PGI2 [11, 12]. Furthermore, ET-1 promotes cell growth, cell adhesion and thrombosis and is increased in lung tissue of patients with pulmonary hypertension (PH). A negative feedback loop is triggered by NO that reduces the affinity of the ETA receptor for ET-1 and can therefore prevent ET-1-mediated signalling [12]. NO can be synthesised by one of three different NO synthases (NOS), of which endothelial (e)NOS is the most important synthase involved in the regulation of the pulmonary vascular tone and is highly expressed in the endothelial cells [13]. NO can bind to its receptor, soluble guanylyl cyclase, which can synthesise the second messenger cyclic guanosine monophosphate, thereby inducing vasodilation. The third important pathway involves prostaglandins and thromboxanes that act on prostanoid receptors which can be divided in receptors that cause relaxation (IP (PTGIR), EP2, EP4 and DP) or contraction (TP, EP1 and FP) of the pulmonary vessels to change the vascular tone [14]. PGI2 is an important mediator of vasodilation which binds and activates the prostaglandin-I2 receptor (PTGIR) [15, 16]. This activation results in vasodilation through the release of cAMP. Apart from these vasoactive factors, retinoic acid signalling has been shown to be important in lung development [17]. Retinoic acid is formed from vitamin A through several sequential enzymatic reactions. Subsequently, it binds to one of the retinoic acid receptors (RAR/RXR) which subsequently modulate transcription of target genes through retinoic acid responsive element [18]. Pharmacological or genetic inactivation of retinoic acid signalling in the early foregut resulted in an increased expression of the Wingless-type MMTV integration site family (Wnt) antagonist Dickkopf (Dkk1) and transforming growth factor (TGF)-β, which plays an important role in airway branching and muscularisation of the pulmonary vasculature [19]. Thus, retinoic acid integrates two important regulatory signalling pathways, Wnt and TGF-β, in the control of Fgf10 expression.

Overview of the major pathways involved in vasodilation and vasoconstriction. ET: endothelin; ECE: endothelin-converting enzyme; ETA: endothelin A; ETB: endothelin B; eNOS: endothelial nitric oxide synthase; sGC: soluble guanylate cyclase; COX: cyclooxygenase; PGIS: prostaglandin synthase; PGI2: prostaglandin I2; AC: adenylate cyclase; TBXAS1: thromboxane synthase; TXA2: thromboxane.
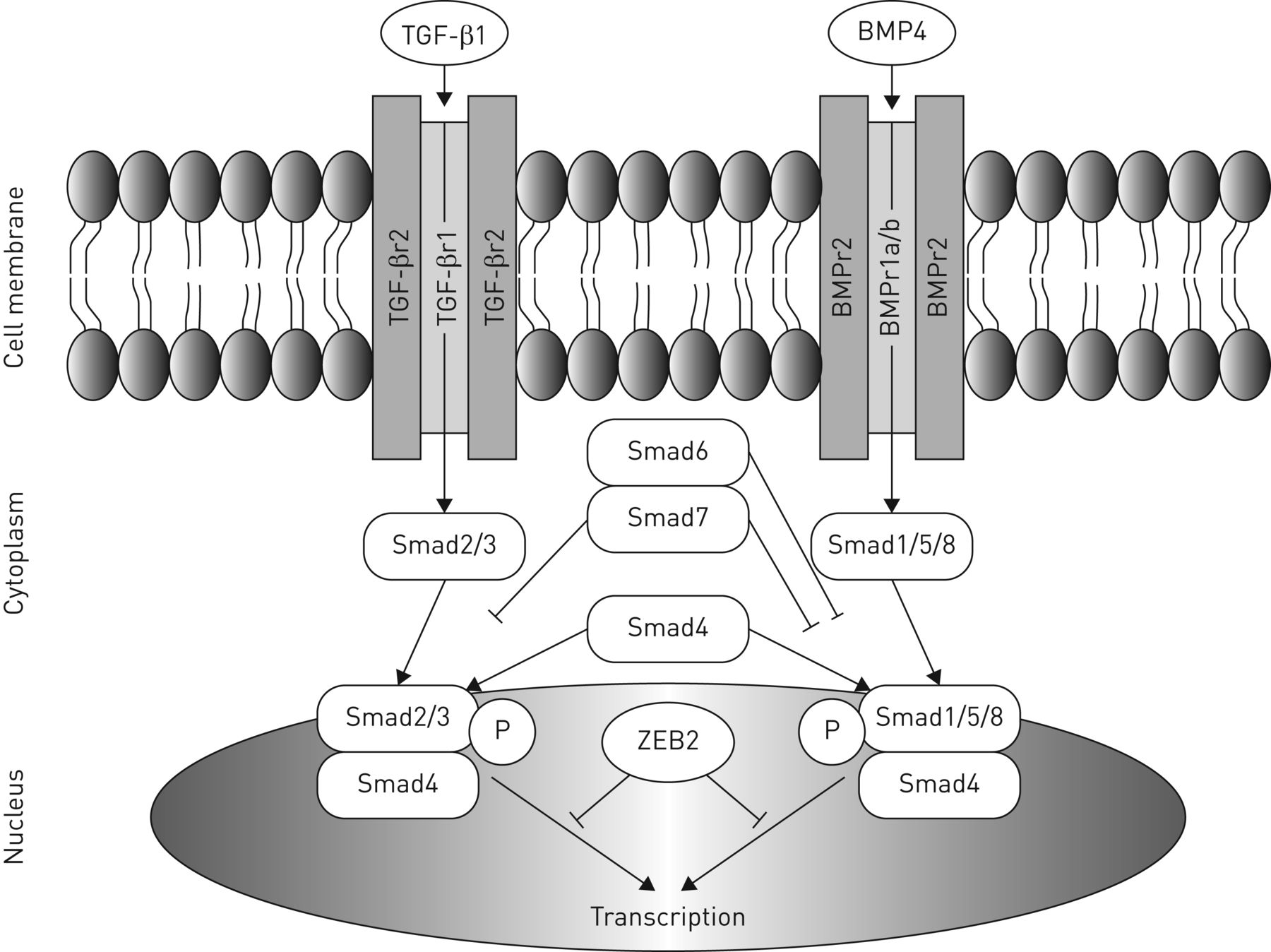
Alveolar formation starts late in development and continues into the first years of life to expand the gas exchanging capacity of the lung. TGF-β partly regulates the concomitant adaptation of the microvasculature. There are two main branches in the TGF-β superfamily: the TGF-β/activin family and the bone morphogenetic protein (BMP) family. All proteins of the TGF-β superfamily act through two classes of serine/threonine kinase receptors: type 1 and type 2. Active TGF-β or BMP can be bound to a specific type 2 receptor, which phosphorylates and activates a type 1 receptor. The activated type 1 receptor subsequently phosphorylates a set of receptor-regulated small body size/mothers against decapentaplegic proteins (Smads), which form a complex with co-Smad (Smad4). TGF-β is responsible for the phosphorylation of Smad2 and Smad 3, whereas BMP phosphorylates Smad 1, 5 and 8. Eventually, the R-Smad/co-Smad complex modulates the transcription of target genes in the nucleus [20] (figure 2). TGF-β signalling is necessary in vascular and airway smooth muscle cells, and in alveolar and airway epithelial cells during late lung development [21]. Previous studies in rodents showed an arrest in alveolarisation both in animals with upregulated TGF-β signalling [22] and after a complete blockade of TGF-β signalling [23], indicating the important differences in TGF-β signalling between early and late lung development.
{kind=link}
{kind=link}
Overview of transforming growth factor (TGF)-β/bone morphogenetic protein (BMP) pathway. ZEB2: zinc finger E-box binding homeobox 2; P: phosphorylation; Smad: small body size/mothers against decapentaplegic proteins
Pulmonary vascular development in congenital diaphragmatic hernia
Congenital diaphragmatic hernia (CDH) is a rare congenital anomaly characterised by a diaphragmatic defect, PH and lung hypoplasia. It has a worldwide incidence of ∼1 in 3000 live births [24, 25] and mortality varies from 20% to 40% [26]. The high mortality and morbidity rates may depend on the presence of associated malformations and/or genetic abnormalities, but are mostly due to the concomitant PH. PH is the result of the altered development of the pulmonary vasculature, the unpredictable vascular reactivity and the disordered process of pulmonary vascular remodelling [27, 28]. PH associated with CDH is characterised by extensive muscularisation of the vessels, which is noticeable early in gestation [29]. This indicates that the structural abnormalities start to develop when the lung is very immature. The pathology of PH is characterised by hypermuscularisation of the midsized and large vessels and neomuscularisation of the small capillaries [18, 28, 30, 31]. Pericytes are prime candidates to underlie and eventually modulate the structural changes observed in PH associated with CDH [6, 32]. Differences in pericyte coverage have been linked to other diseases with a prominent vascular component, such as diabetic retinopathy, cancer and adult pulmonary arterial hypertension [33–35]. Additionally, co-cultured pulmonary arterial endothelial cells and smooth muscle cells showed an altered interaction when cells were used from a CDH sheep model [36].
Aberrations in the expression of different factors in the important vasoactive pathways have previously been described by us and others. Increased levels of both the ETA and ETB receptors were shown in human CDH [37] and high plasma levels of circulating ET-1 were associated with the severity of PH in this population [38]. Studies on the expression of eNOS showed variable results, where a decrease was found in some human studies [39], but no differences or even an increased expression of eNOS was shown by us and others [40]. No studies have been performed on the expression of factors of the PGI2 pathway.
Finally, recent studies implicated microRNAs in the development of CDH, but it remains to be seen whether the changes observed in miR-200b and miR-10a are a cause or a consequence of CDH [41, 42].
Treatment of pulmonary vascular defects in CDH
The significance of PH in the mortality and morbidity of patients with CDH has been increasingly recognised. Extracorporeal membrane oxygenation may be provided as a treatment of last resort to relieve the respiratory distress of these children and provide time for the pulmonary vasculature to adjust to the postnatal increased flow to the pulmonary vascular bed. However, the persistent PH requires more targeted approaches to improve the oxygenation of these children. The first vasodilators used in these critically ill newborns with PH, tolazoline [43], an α-adrenergic receptor antagonist, and prostacyclin [44], a PGI2 agonist, resulted in variable results. Tolazoline infusion showed a response in 21 out of 36 neonatal patients with lung disease, where systemic hypotension and bleeding tendency were seen as a severe side-effect in several patients [43]. Treatment with PGI2 improved pulmonary arterial pressure and oxygenation in two out of three neonatal patients with idiopathic PH and PH caused by meconium aspiration. However, it had no beneficial effect in two patients with CDH, whereas systemic hypotension was seen as a side-effect in one of these patients [44]. Currently, patients with CDH still respond unpredictably to the available vasodilator therapy due to the lack of understanding of the underlying mechanisms in the individual patient. Inhaled (i)NO is most commonly used, but studies have failed to show its efficacy in this specific group of patients [45]. Apart from iNO, sildenafil and some prostaglandin analogues are used as rescue therapy in a compassionate way in the most severe cases, but with variable results [46–48]. No appropriate trials have been performed on these drugs and, with a few exceptions, no data on pharmacokinetics are available for CDH neonates. In the near future a comparative effectiveness trial will be conducted to compare iNO with i.v. sildenafil under the guidance of the CDH-EURO Consortium.
Currently, the only prenatal intervention used in CDH undergoing clinical trial is fetoscopic endoluminal tracheal occlusion (FETO), where a small balloon is inserted into the fetal trachea to temporarily block the airway for a period of 4–6 weeks (Tracheal Occlusion to Accelerate Lung Growth (TOTAL); clinicaltrials.gov NCT02875860) [49]. As a result, fluid is trapped in the lungs, creating internal pressure which forces the lung to grow. This idea of blocking the airway emerged from an experiment of nature in patients with congenital high airway obstruction syndrome resulting in big polyalveolar lungs [50]. Previous research has shown that tracheal occlusion can indeed cause an increase in lung growth [51, 52] and removing the balloon before birth has shown to be necessary for a better maturation of the lung by decreasing the apoptosis of the alveolar type 2 cells, which produce surfactant, an essential compound for lung function [53]. So far, FETO has been shown to improve survival rate in high-risk CDH patients, but at the cost of increased morbidity and premature delivery [54–56]. The results of the TOTAL trial will hopefully provide a more definitive answer.
Currently, treatment with vasodilators in patients with CDH is only used postnatally, where previous research has shown already major differences in the pulmonary vasculature early in development [29]. We and others have performed some studies on the antenatal use of the phosphodiesterase-5 inhibitor sildenafil in different animal models of CDH, showing improvement in alveolarisation and pulmonary vascular development [57–61]. However, the pulmonary pathology in these treated animals did not show complete reversal to the normal lung histology.
The absence of an integrated analysis of the hypermuscularisation, pericyte coverage and the pathways regulating vascular tone, as well as the ineffective postnatal treatment in individual patients with CDH makes it essential to obtain greater insight into the aberrant development of the pulmonary vasculature to optimise treatment in these patients. Furthermore, adequate prenatal treatment could be beneficial for pulmonary development and its vascular pathology, in particular in newborns with CDH.
Footnotes
Support statement: This work was supported in part by the Sophia Foundation for Medical Research grant number 678 (awarded to H.M. Kool). Funding information for this article has been deposited with the Crossref Funder Registry.
Conflict of interest: None declared.
Provenance: Commissioned article, peer reviewed.
- Received September 7, 2017.
- Accepted November 9, 2017.
- Copyright ©ERS 2018.
ERR articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










