





Abstract
Children's interstitial lung diseases (chILD) are increasingly recognised and contain many lung developmental and genetic disorders not yet identified in adult pneumology. Worldwide, several registers have been established. The Australasian Registry Network for Orphan Lung Disease (ARNOLD) has identified problems in estimating rare disease prevalence; focusing on chILD in immunocompetent patients, a period prevalence of 1.5 cases per million children and a mortality rate of 7% were determined. The chILD-EU register highlighted the workload to be covered per patient included and provided protocols for diagnosis and initial treatment, similar to the United States chILD network. Whereas case reports may be useful for young physicians to practise writing articles, cohorts of patients can catapult progress, as demonstrated by recent studies on persistent tachypnoea of infancy, hypersensitivity pneumonitis in children and interstitial lung disease related to interferonopathies from mutations in transmembrane protein 173. Translational research has linked heterozygous mutations in the ABCA3 transporter to an increased risk of interstitial lung diseases, not only in neonates, but also in older children and adults. For surfactant dysfunction disorders in infancy and early childhood, lung transplantation was reported to be as successful as in adult patients. Mutual potentiation of paediatric and adult pneumologists is mandatory in this rapidly extending field for successful future development.
This brief review highlights publications in the field of paediatric interstitial lung disease as reviewed during the Clinical Year in Review session presented at the 2017 European Respiratory Society (ERS) Annual Congress in Milan, Italy. It was commissioned by the ERS and critically presents progress made as well as drawbacks.
Abstract
Successful developments in chILD are register/consortia based and potentiate paediatric and adult pneumology http://ow.ly/dgrO30hBbRJ
Definitions
The name interstitial lung disease (ILD) is used interchangeably with diffuse parenchymal lung disease (DPLD). The term “interstitial” may be misleading, as it appears to be too narrow when used strictly, because other parenchymal components such as vessels, epithelium, airways or pleura are usually also involved. For convenience and propagation, the acronym chILD (children's ILD) is used to differentiate from adult ILD. According to the clinicopathological and genetic classification system, groups of diseases predominantly occurring in infancy are labelled “A”, whereas those disorders manifesting at all ages are labelled “B” (table 1) [1, 2].
Results of PubMed search for “interstitial lung disease in children” between June 2016 and June 2017
chILD research performance lags behind adult ILD: time to change
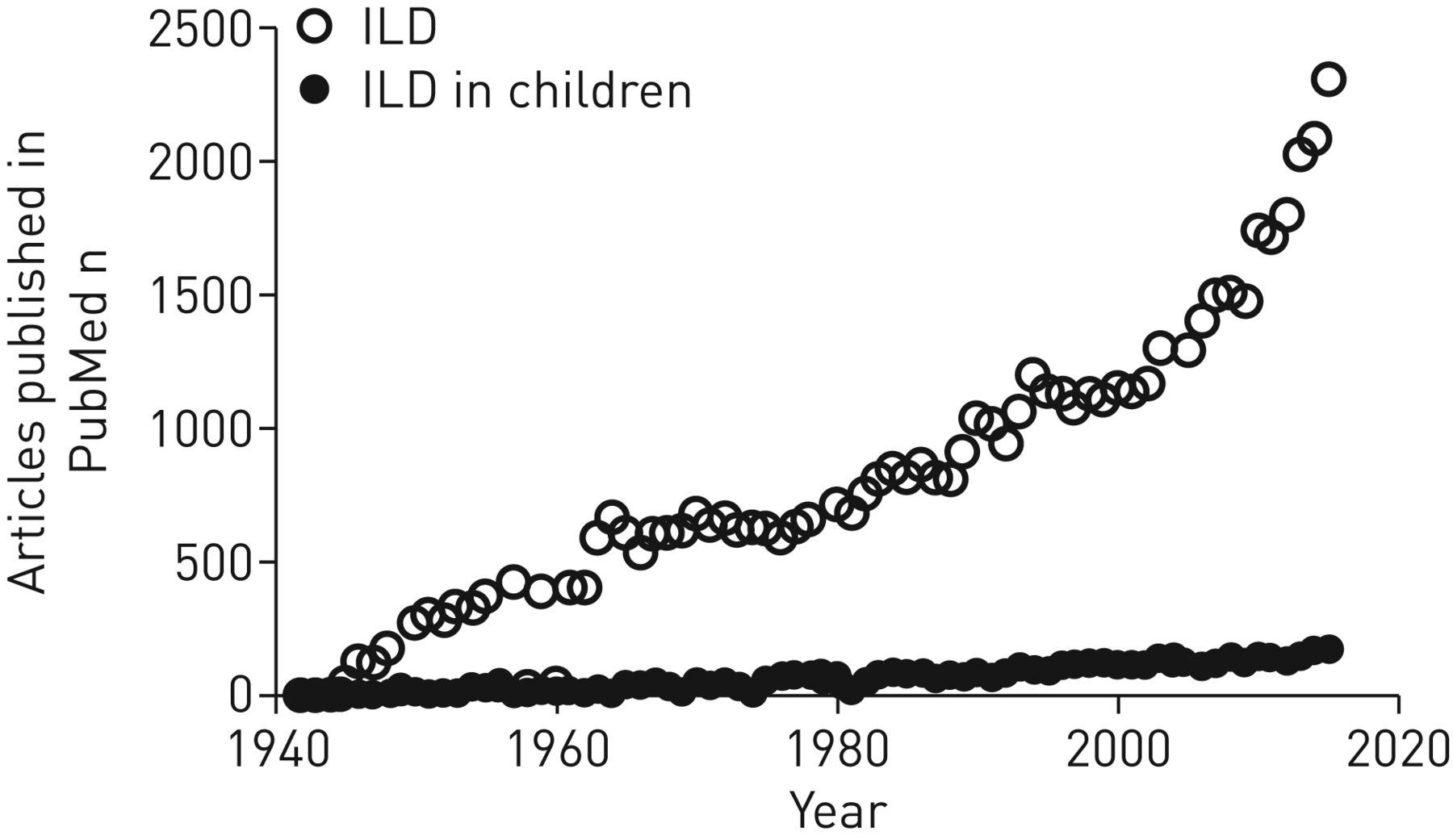
chILD/DPLD in children [1] represents an underdeveloped area of pulmonary research, illustrated by comparing the numbers of publications on ILD in adults and those in chILD (figure 1). Approximately 100 times more publications are referenced for adult ILD. The reasons may include the higher incidence and prevalence of adult cases over the lifetime [2, 3]. However, research efforts in adult ILD and resulting publications have increased in the past decade, but this was not the case for chILD (figure 1). Beyond the similarity of chILD phenotypes to other, more common lung diseases, and resulting difficulties of identification; the geographically wide scattering of cases; and that many diseases have not yet been described at all, other reasons must be of significance. These include a lack of a critical mass of both identified cases and centres with expertise. Interested physicians, who identify and publish individual cases “which are just another one of the same” invest a lot of effort, enthusiasm and money with little effect. This is illustrated by a search using PubMed and the search term “interstitial lung disease in children”. Between June 2016 and June 2017 148 articles were published (table 1). Of these, more than half were case reports and reviews mainly on case reports. Only a few manuscripts collected series of patients to describe the entity or treatments. A role model for handling rare cases of diseases comes from paediatric oncology [4]. The results of this PubMed search illustrate this, showing that due to the presence of “interstitial lung disease” in pulmonary histiocytosis, 16 out of the 19 reports retrieved in the category “DPLD related to systemic disease processes” were linked to this entity (table 1), whereas the other entities had much lower numbers. The presence of monogenetic disease entities that primarily manifest in infancy [5–8] are a unique opportunity to develop comprehensive disease-categorisation systems, to extend the database of molecularly defined pulmonary disease entities [9] and to collaborate in all these aspects with pneumologists, radiologists, pathologists and geneticists [10] for a personalised diagnostic and therapeutic approach.

Annual numbers of publications retrieved by PubMed (searched in June 2017) with the term “interstitial lung disease” (ILD) for the age groups ≤18 years (ILD in children) and >18 years.
Structuring advances in chILD by consortia-run registers and biobanks
A promising approach to solve these problems is to collect cases and biomaterials in registers. Whereas in the past overreaching efforts to collect cohorts to study the natural course, molecular disease causes, appropriate diagnostic steps, treatment options and meaningful prophylaxis were undertaken infrequently, in recent years several national and international registers have been established [3, 11–14]. A few databases exist, in particular the international chILD-EU register, the national French RespiRare and the Australasian Registry Network for Orphan Lung Disease (ARNOLD) for adult and paediatric rare lung diseases. In 2016 ARNOLD reported their prevalence data between 2009 and 2014 [15]. Although the system worked comfortably via email, under-reporting was a major problem, as only 32% of the physicians approached responded. This is a problem faced in many other areas of clinical research into rare diseases. A major conclusion from all these studies is that it is naïve to expect that the effort necessary to include subjects in any register can be achieved without providing concrete support and clear incentives. Engagement in registries for single diseases such as idiopathic pulmonary fibrosis or cystic fibrosis is superior, probably related to the availability of research coordinators entering data and shipping biomaterials [15]. The workload to enter a case is ∼5 h, and includes obtaining consent, collection of clinical data, burning of radiology imagines onto compact discs, collecting pathology specimens and biomaterials, shipping to a central site and writing a case description [16].
Focusing on chILD in immunocompetent patients, paediatric pulmonologists in Australia and New Zealand and caring for patients aged 0–18 years completed a questionnaire on demographics, clinical features and outcomes, over a 10-year period. These data, together with data from the two reference genetics laboratories, were used to calculate prevalence [17]. 115 cases were identified, yielding a period prevalence of 1.5 (range 0.8–2.1) cases per million children. Two-thirds of the patients were aged <2 years. Overall clinical outcome was good in 79% of patients; the mortality rate was 7% [17].
These collaborations are fruitful to improve definitions, such as for exacerbations. The chILD-EU collaboration conducted a systematic literature review, followed by a two-round online Delphi process to define and propose criteria for an acute exacerbation in chILD [18]. A definition of acute exacerbations in chILD was accepted and a set of diagnostic criteria was selected, based on expert knowledge (table 2). It was proposed that at least two of the criteria listed must be met. Using this framework, the reliability, validity, reproducibility and age-dependent applicability can be assessed in prospective studies of children, as well as their families.
Acute exacerbation in children's interstitial lung disease
Disease cohorts can catapult progress compared to repetitive cases
Case reports represent an important tool for young physicians gain experience in writing articles and to enter the academic arena. However, they are inferior in generating systematic knowledge on diseases to move the field forward. Cohorts of rare diseases are useful for the description of their manifestation, course and outcome; they can help obtain diagnostic markers, establish pathophysiology and treatments. As they age, surviving patients with diseases characteristic of infancy reach adulthood and as cohorts they can be followed with great advantage to determine the natural course of the disease. A very important issue will be the transition from paediatric to adult care. To achieve this successfully, early training and involvement of teams primarily caring for adult patients with ILD will be of key importance.
Based on the pathological classification system first introduced for children aged <2 years and based on lung biopsies by Deutsch et al. [19], a clinicopathological and genetic classification system was established and validated [1, 2]. This system has the advantage of accommodating cases not diagnosed by biopsy, those with inconclusive diagnosis or some lacking information. For example, there are many children of all ages with respiratory distress syndrome who cannot reliably be allocated, either due to lack of diagnostic procedures or who remain inconclusive even after all diagnostic means available have been explored. For these cases defined categories are available; these can be revisited later as more advanced diagnostic techniques become available. Entities that can newly be diagnosed genetically, such as pulmonary hypertension caused by mutations leading to capillary haemangiomatosis/veno-occlusive disease or the interferonopathies (see later), are accommodated. As the main purpose of this system is greatest possible grouping of similar diseases to build cohorts, constant refinement in subcategories is in progress. The system is widely used by the chILD-EU management platform (www.childeu.net) and the reliability and precision of categorisation has been assessed [14, 16]. The publications reported in this review are ordered according to this classification system.
DPLD group A3: infant conditions of undefined aetiology
Persistent tachypnoea of infancy
An important phenotype of ILD in infants comprises a persistent tachypnoea, crackles on auscultation, failure to thrive and borderline oxygen saturation. The condition is rare and often remains undiagnosed, as the phenotype is variable over time and often relatively mild. Importantly, it must be differentiated from chronic forms of surfactant dysfunction disorders.
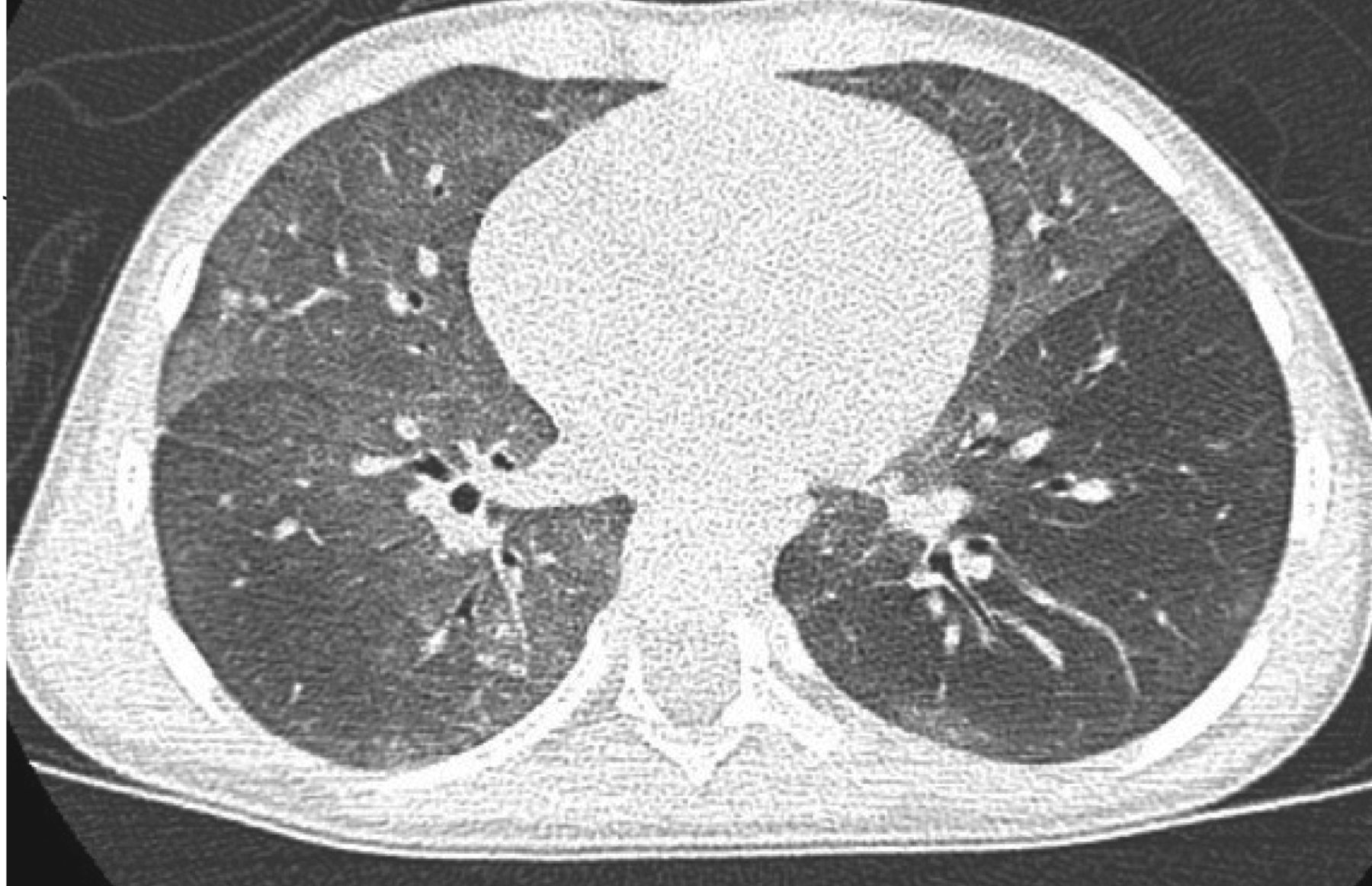
Rauch et al. [20] collected a cohort of 89 such infants, after the exclusion of other conditions, including surfactant dysfunction disorders, immune deficiencies, cystic fibrosis, primary ciliary dyskinesia and cardiac disease. This condition is called persistent tachypnoea of infancy (PTI). An important diagnostic evaluation is computed tomographic (CT) scanning, characteristically yielding ground-glass attenuation in typical locations, including the middle lobe, lingula, paramediastinal or parahilar lung segments (usual PTI) (figure 2), or having ground-glass opacities in atypical locations or having additional minor abnormalities (aberrant PTI). They found that the age of onset of symptoms (3.5 (range 0–20) months), the initial symptoms (tachypnoea 100%, crackles 86%, retractions 82%, failure to thrive 66% and hypoxaemia 88%) were not different between usual and aberrant PTI. After an average follow-up until 4.2 (range 0.7–10.5) years, 50% of the infants were healthy, 46% were still sick, but had improved, and only 4% were in the same clinical condition [20]. It is of great importance to know and recognise this condition, in order to avoid overtreatment or to miss treatments necessary for similar conditions. The differential diagnosis is less simple, particularly because the underlying conditions are probably a collection of diseases with different histological features and possibly genotype. In those 33 subjects of the cohort with lung biopsies, only 21 had neuroendocrine cell hyperplasia of infancy (NEHI). Four infants had pulmonary interstitial glycogenosis (PIG) and eight children had completely normal histology with no increase in neuroendocrine cells. This may be due to sampling error or lack of increased neuroendocrine cells in these patients. Thus, lung biopsies may be helpful in rare and complicated cases, but are not necessary in all infants, given that a very close and long-term observation is guaranteed.

{kind=link}
{kind=link}
Ground-glass opacity in lingular, middle lobe and parahilar areas on inspiratory scan in an infant with persistent tachypnoea of infancy, usual form and biopsy-proven neuroendocrine cell hyperplasia.
Houin et al. [21] describe exacerbations in two infants with PTI, one of them previously having had a lung biopsy showing increased bombesin-positive cells. These exacerbations were observed at 5 and 6 years and were characterised by increased air-trapping, demonstrated by lung function and CT imaging. The authors suggest that such exacerbations may be different from those seen in asthma or normal children with obstructive bronchitis [21]. This leads to the difficult issue of acute exacerbations in chILD as reported earlier; obviously a prospective and multinational research effort would be the most promising to evaluate this issue in much larger cohorts of infants.
Diseases in this category comprehend infants with a core clinical phenotype of persistent tachypnoea and characteristic CT imaging with ground glass. After appropriate differential diagnosis and without biopsy, conditions are currently collected under the label PTI. If clinically indicated, a lung biopsy may differentiate other entities, including NEHI and PIG. In a few of these cases, genetic analysis has diagnosed NKX-2.1 [22] or FOXP1 [23] deficiencies, and others likely are to follow.
DPLD group A4: ILD related to alveolar surfactant region
Rare mutations in the surfactant dysfunction genes and in particular ABCA3 have been shown to lead to respiratory distress syndrome in the mature neonate and to ILD in children and adults of European descent in the United States [24]. These variations were either present in a homozygous or compound heterozygous arrangement, as expected for an autosomal recessive condition. In addition, heterozygous mutations in ABCA3, such as R288K have been associated with increased rates of neonatal respiratory distress [25] or ILD in older children [26]. In a retrospective cohort study of 228 children with ILD related to the alveolar surfactant system, nine children with ILD carrying a single (heterozygous) R288K variant, a frequency significantly higher than in the general Caucasian population, were found. At birth, all had neonatal respiratory distress, recovered and developed chronic ILD with intermittent exacerbations during early childhood. Detailed analysis of the R288K variant in ABCA3 showed smaller lamellar bodies and a reduced dipalmitoyl-phosphatidylcholine content and decreased lamellar body volume of the cells studied [26]. It was suggested that the heterozygous R288K variant can underlie ILD in childhood as a significant risk factor.
Of interest are the rather high carrier rates for ABCA3 mutations of 3–5%, yielding frequencies of ABCA3 deficiency at ∼1 in 3100 individuals of European descent. Such frequencies are similar to those for cystic fibrosis (1 in 2500 infants of European descent) [25]. Chen et al. [27] investigated the carrier rate in Chinese populations and found it to be 1.3%, which was significantly lower than expected from previous data. The authors conclude that the known high rate of respiratory distress in the Chinese population must be due to other genes or factors affecting the alveolar type 2 cell metabolism or lung development. Unfortunately, no such data are yet available for Europe.
Lung transplantation may be a treatment option for infants and young children with genetic disorders of surfactant metabolism, including SFTPB, SFTPC, ABCA3 and NKX2-1 deficiency. Although rarely performed in Europe, such transplants have been undertaken at St Louis Children's Hospital (St Louis, MO, USA) for more than two decades. Between 1993 and 2015, 44 children were reported; 28 of these were transplanted at age <1 year, and 16 were older. As expected from disease manifestations, infants underwent transplantation more frequently for SFTPB deficiency, compared to children for SFTPC mutations. ABCA3 deficiency was more evenly distributed over age. Infants who received an organ earlier were more likely to be mechanically ventilated at the time of transplantation, had more speech and motor delays, but were less likely to develop bronchiolitis obliterans post-transplantation. Despite advances in immunosuppressive, supportive respiratory and nutritional therapies, mortality did not differ between infants and children or between the earlier (1993–2003) and the later (2004–2015) epochs [28].
DPLD group B2: ILD related to exposures in the immune intact patient
Hypersensitivity pneumonitis
Hypersensitivity pneumonitis, also called exogenous allergic alveolitis in Europe, represents ∼50% of chILD [1, 29] and is thus an important entity, as it may lead to chronic and severe lung disease if not recognised [30]. Sisman et al. [31] performed a cross-sectional follow-up study in patients previously diagnosed with biopsy and high-resolution CT-verified hypersensitivity pneumonitis during childhood. A broad array of diagnostic tools was used to characterise the patients, who were followed for a median 3.3 (range 0.2–10.9) years. Of the 22 patients assessed, the lung clearance index was abnormal in 47%, in contrast to abnormal forced expiratory volume in 1s (FEV1) in only 9%. Obstruction was fixed, as there was no significant bronchodilator responsiveness. Diffusing capacity of the lung for carbon monoxide (DLCO) and total lung capacity were abnormal in 41% and 14%, respectively. During standardised exercise testing, only 11% demonstrated abnormal maximal oxygen uptake. Importantly, there was no change of outcome measures between end of treatment and end of time of follow-up. The authors concluded that long-term prognosis appears to be favourable, although persistent peripheral airway involvement was present in about half of the subjects. Further investigations and follow-up are necessary to determine its clinical significance.
DPLD group B3: ILD related to immune dysregulation or compromise
Interferonopathies initially manifesting solely with pulmonary disease
An inherited autoinflammatory syndrome with the main clinical features onset in infancy, systemic inflammation, cutaneous vasculopathy, pulmonary inflammation and caused by mutations in the transmembrane protein 173 (TMEM173) has been described [32, 33] and named SAVI (stimulator of interferon genes-associated vasculopathy with onset in infancy). Of great importance is the article by Picard et al. [34], demonstrating a pulmonary-only phenotype of this syndrome manifesting during childhood.
A child investigated at age 5 years for clubbing and chronic cough had failure to thrive since the first year of life. His mother had ILD with FEV1/forced vital capacity of 50% predicted, which had been followed since the age of 20 years, i.e. had experienced undiagnosed pulmonary problems earlier. Another infant had failure to thrive and chronic pulmonary problems, which were initially related to severe dysphagia and gastro-oesophageal reflux, but persisted despite of treatment of reflux and discontinuation of oral feeding. At 3 years dyspnoea with exercise and hypoxaemia appeared progressively. Systemic symptoms were not reported; at 7.5 years restrictive lung function and decreased DLCO were noted. On CT scans ground glass, cysts, reticulation, hyperinflation, traction bronchiectasis and lymphadenopathy developed over time. Abnormal immunological tests (decreased CD4 T-cells, natural killer lymphocytes and increased IgG and IgA) and slightly positive antinuclear antibodies were present. Lung biopsies showed lymphocytic inflammatory infiltrates. Known genes diseases causing pulmonary fibrosis (SFTPC, SFTPB, ABCA3, TTF1, TERT and TERC) were excluded. This serves as an example to illustrate that many conditions caused by immune dysregulation or compromise can presenting with chronic pulmonary features. As these are ultra-rare conditions, even rather small groups of subjects may be sufficient for successful deduction of genetic aetiology.
Conclusion
Among patients with diffuse parenchymal lung diseases, chILD is heavily under-recognised regarding clinical care and translational and patient-centered research activities. The contribution of cases to worldwide registers is important, to go beyond individual case reports which frequently have not much novelty. Cohort studies on rare diseases such as PTI, hypersensitivity pneumonitis in children and ILD related to interferonopathies from mutations in the transmembrane protein 173 are vital for progress to be made. Novel, mostly rare, disease entities are increasingly diagnosed in paediatric patients. Pneumologists will have to prepare for and meet the needs of these young adults as they transfer into adult medicine.
Acknowledgement
This review is based on PubMed search results using the search term “interstitial lung disease in children” and the papers retrieved. The author apologises to researchers whose frequently excellent papers and reviews were not included due to limited space.
Footnotes
Support statement: The work was supported by chILD-EU (FP7 No. 305653), the Bundesministerium für Bildung und Forschung (BMBF), Deutschland, project “HCQ4Surfdefect”, under the frame of E-Rare-3, the ERA-Net for Research on Rare Diseases and the European Cooperation in Science and Technology COST A16125. Funding information for this article has been deposited with the Crossref Funder Registry.
Conflict of interest: None declared.
Provenance: Commissioned article, peer reviewed.
- Received August 31, 2017.
- Accepted November 24, 2017.
- Copyright ©ERS 2018.
ERR articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References











Jump To
- Article
- Abstract
- Abstract
- Definitions
- chILD research performance lags behind adult ILD: time to change
- Structuring advances in chILD by consortia-run registers and biobanks
- Disease cohorts can catapult progress compared to repetitive cases
- DPLD group A3: infant conditions of undefined aetiology
- DPLD group A4: ILD related to alveolar surfactant region
- DPLD group B2: ILD related to exposures in the immune intact patient
- DPLD group B3: ILD related to immune dysregulation or compromise
- Conclusion
- Acknowledgement
- Footnotes
- References
- Figures & Data
- Info & Metrics