





Abstract
Dyspnoea is a principal presenting symptom in pulmonary arterial hypertension (PAH), and often the most distressing. The pathophysiology of PAH is relatively well understood, with the primary abnormality of pulmonary vascular disease resulting in a combination of impaired cardiac output on exercise and abnormal gas exchange, both contributing to increased ventilatory drive. However, increased ventilatory drive is not the sole explanation for the complex neurophysiological and neuropsychological symptom of dyspnoea, with other significant contributions from skeletal muscle reflexes, respiratory muscle function, and psychological and emotional status. In this review, we explore the physiological aspects of dyspnoea in PAH, both in terms of the central cardiopulmonary abnormalities of PAH and the wider, systemic impact of PAH, and how these interact with common comorbidities. Finally, we discuss its relationship with disease severity.
Abstract
Dyspnoea is a complex integration of all the cardiopulmonary and systemic abnormalities in PAH http://ow.ly/D13W30dMDwJ
Introduction
Pulmonary arterial hypertension (PAH) is a primary disease of the pulmonary vasculature, which manifests physiologically principally through increased afterload on the right ventricle causing right ventricular failure, first through failure to augment cardiac output on exercise to match the exercise workload and subsequently, at rest, leading to progressive debilitating symptoms and death [1]. PAH is the quintessential cardiopulmonary disease, exerting its effects through both decreased cardiac output and impaired gas exchange, but also has far-reaching systemic effects on multiple organs through various mechanisms, including decreased tissue perfusion, inflammation and possible paraphenomena, such as autoimmunity [2].
On the face of it, dyspnoea may be considered to be a straightforward symptom, but this is by no means the case. The first descriptions of “cardiac dyspnoea” in the medical literature date back to 1895. Loomis [3] proposed in a definition of this phenomenon that “the difficulty of breathing is due entirely to a cardiac condition” with “no organic changes within the lungs, bronchial tubes, or larynx to obstruct the entrance of air to the alveolar surfaces.” Interestingly, he describes that autopsies of patients who fulfilled this definition and died showed that “usually the arrest is in the right heart, and as a result the blood is shut off from the pulmonary artery. The lungs under such circumstances will be not only free from congestion but more or less bloodless, while the other internal organs will be found intensely congested.”
Most cases of PAH on an incremental exercise test are physiologically limited by cardiovascular function and not by lack of breathing reserve, yet the cardinal presenting feature of PAH is dyspnoea; once diagnosed and treated this is also perhaps the most dominant everyday symptom of PAH [4].
It is therefore important to consider that while patients may describe dyspnoea during everyday activities and often as the limiting factor in their activities, when undertaking an incremental test to maximum effort, other features come into play. Typical cardiovascular symptoms would include leg fatigue and presyncope or syncope. Although dyspnoea can be quantified by scores, such as the Dyspnoea-12 score [5], when the term “breathlessness” is used by PAH patients to describe their symptoms, it is a more complex summation of all the physiological disturbances in PAH. Dyspnoea may also be of prognostic relevance, with self-reported dyspnoea being identified as an independent predictor of the risk of death from cardiac causes in patients referred for cardiac stress testing [6].
There is a paucity of studies examining the specific mechanisms of dyspnoea in PAH. As a result, in this review, we have taken the approach to examine the individual components of exercise limitation in PAH and how they feed into dyspnoea, as well as describing how they contribute to the overall reduction in exercise capacity.
Cardiovascular response to exercise in PAH
The cardiovascular system responds to exercise in order to process the metabolic changes that occur with increased muscle work, including: 1) an increased, workload-dependent oxygen demand in the skeletal muscles, and 2) an increased generation of metabolic products affecting acid–base homeostasis, such as carbon dioxide.
According to the Fick principle, cardiac output can be calculated if oxygen uptake (V′O2) and the arterial–mixed venous oxygen content difference (Ca–vO2) are known: In order to identify the cardiovascular components that determine an increase in V′O2, the Fick equation may be rearranged and expanded:
In order to identify the cardiovascular components that determine an increase in V′O2, the Fick equation may be rearranged and expanded: This equation shows that changes in V′O2 are directly dependent on three parameters: stroke volume, heart rate and the Ca–vO2. These parameters themselves reflect additional physiological adaptative mechanisms, with stroke volume depending on systolic and diastolic ventricular function and valvular competency, and heart rate depending on chronotropic competence and stimulation of adrenergic receptors. The Ca–vO2 reflects oxygen transport capacity (haemoglobin) and peripheral oxygen extraction in the peripheral muscles. Therefore, V′O2 is a comprehensive but nonspecific measure of cardiovascular and metabolic function and correlates with haemodynamic changes. However, this equation also demonstrates that any increase in V′O2 will be equal to the increase in the product of systemic oxygen extraction (Ca–vO2) and cardiac output (heart rate×stroke volume). Both mechanisms may be pathologically altered in PAH.
This equation shows that changes in V′O2 are directly dependent on three parameters: stroke volume, heart rate and the Ca–vO2. These parameters themselves reflect additional physiological adaptative mechanisms, with stroke volume depending on systolic and diastolic ventricular function and valvular competency, and heart rate depending on chronotropic competence and stimulation of adrenergic receptors. The Ca–vO2 reflects oxygen transport capacity (haemoglobin) and peripheral oxygen extraction in the peripheral muscles. Therefore, V′O2 is a comprehensive but nonspecific measure of cardiovascular and metabolic function and correlates with haemodynamic changes. However, this equation also demonstrates that any increase in V′O2 will be equal to the increase in the product of systemic oxygen extraction (Ca–vO2) and cardiac output (heart rate×stroke volume). Both mechanisms may be pathologically altered in PAH.
The predominant cardiovascular abnormality of PAH on exercise is that of a failure to augment cardiac output on exercise due to increased right ventricular afterload, and this in turn is largely due to a failure to augment cardiac stroke volume [7]. Unlike in healthy subjects, where pulmonary vascular resistance falls on exercise due to pulmonary vascular recruitment and distensibility of the resistive vessels [8], the pulmonary vascular resistance does not fall in PAH to accommodate rises in cardiac output and pulmonary arterial pressure. Thus, increases in pulmonary arterial pressure on exercise reflect changes in cardiac output and stroke volume. Such a limitation in right ventricular contractile reserve during exercise may lead to impaired left ventricular filling during exercise and, as a consequence, exercise systemic blood pressure may not increase adequately, leading to presyncope and syncope in patients with advanced disease. In fact, several studies have been able to demonstrate that this aggregate of pathophysiological abnormalities contains relevant prognostic information. The absence of an adequate right ventricular contractile reserve [9] as well as the failure to increase systemic blood pressure during exercise [10] have been identified as predictors of poor prognosis, whereas greater increases in exercise cardiac output and systolic pulmonary arterial pressure identify better prognosis and response to therapy [7, 11].
Skeletal muscle dysfunction and severe, chronic deconditioning may be an underestimated problem in PAH patients [12]. With respect to the Fick equation, skeletal muscle function during exercise relates to Ca–vO2, as remaining organ oxygen demand is assumed to be relatively constant during exercise, and an increase in oxygen extraction is mainly caused by increased skeletal muscle metabolism. However, oxygen extraction is difficult to measure in clinical practice, as it requires sampling of mixed venous blood. Therefore, a direct evaluation of skeletal muscle function in clinical practice, outside of studies, is difficult.
Only limited data are available regarding the Ca–vO2 during exercise in pulmonary hypertension. In one study, maximum systemic oxygen extraction during exercise was impaired in PAH, compared with pulmonary venous hypertension, suggesting that this may contribute to exercise intolerance [13]. However, this is in contrast to studies of heart failure (which would be expected to share many similarities with PAH) that showed an increase in peak oxygen extraction similar to that found in highly trained athletes [14]. Further studies in heart failure have demonstrated that impaired diffusional transport of oxygen may be amenable to an exercise rehabilitation programme [15], but a recent detailed assessment of the exercise haemodynamic changes following a comprehensive exercise programme did not report on these aspects [16]. Impairment of muscle flow has not been evaluated in pulmonary hypertension, but may be an additional contributing factor, as previously demonstrated in heart failure patients [15, 17].
This combination of cardiac impairment and skeletal muscle dysfunction leads to impaired oxygen delivery and oxygen extraction, resulting in early anaerobiasis, and this in turn results in increased carbon dioxide production (V′CO2), necessitating an increase in alveolar ventilation. It is thus relatively straightforward to understand how these components of the “PAH syndrome” result in an increased ventilatory demand. However, the link to the sensation of dyspnoea is not so clear. While ventilation remains coupled to V′CO2 in order to maintain pH, as a result of early anaerobic metabolism, its relation to V′O2 and work will become uncoupled. The sensation of ventilation being out of proportion to workload is potentially one of the mechanisms by which dyspnoea is perceived [18]. This may be mediated, at least in part, by afferent muscle reflexes themselves, the so-called “metaboreflex” and “mechanoreflex”, which feed into central pathways regulating the cardiovascular and ventilatory responses to exercise as well as the sensation of effort [19]. These reflexes in turn integrate with other afferent inputs, e.g. chemoreceptors and respiratory afferents, to produce the sensation of dyspnoea (figure 1). This is beyond the scope of this review and has been reviewed in more detail elsewhere [18, 20].

Schematic representation of some of the inputs into the sensation of dyspnoea.
Lastly, patients may experience myocardial ischaemia, further contributing to the symptoms of dyspnoea. Usually, this relates to an imbalance between oxygen supply and demand, but in rare circumstances pulmonary artery dilatation may cause a significant compression of the left coronary artery main stem (figure 2) and potentially lead to unexpected sudden death in precapillary pulmonary hypertension [21].

Computed tomography coronary angiogram from a 43-year-old female patient with pulmonary arterial hypertension, showing compression of the left coronary artery main stem by a large pulmonary artery (PA) aneurysm. LCA: left coronary artery; AOV: aortic valve.
Respiratory response to exercise in PAH
The ventilatory response to exercise in PAH can be considered as an interaction between ventilatory demand, ventilatory pump function, breathing pattern and indirect inputs such as effort–output uncoupling and emotion. These are summarised in figure 1. In an incremental exercise test to maximal effort, the ventilatory demand, or more simply, ventilation, is considered in relation to V′CO2 and is driven by a number of factors, all of which are altered in PAH, resulting in ventilatory inefficiency. The relationship between minute ventilation (V′E), V′CO2 and its other modifiying factors can be expressed as follows: PaCO2 is the arterial partial pressure of carbon dioxide and VD/VT is the physiological dead space volume as a fraction of tidal volume. As a result of earlier anaerobiasis, V′CO2 is increased earlier in exercise in PAH than in healthy individuals due to the buffering of lactic acid, leading to increased ventilatory demand at lower workloads:
PaCO2 is the arterial partial pressure of carbon dioxide and VD/VT is the physiological dead space volume as a fraction of tidal volume. As a result of earlier anaerobiasis, V′CO2 is increased earlier in exercise in PAH than in healthy individuals due to the buffering of lactic acid, leading to increased ventilatory demand at lower workloads: In PAH, PaCO2 is reduced due to chronic hyperventilation, which is thought to relate to heart failure and increased chemosensitivity [22–24]. This increases the ventilatory demand in relation to carbon dioxide output, already increased through early anaerobiasis. This is often referred to as ventilatory inefficiency and quantified as the ratio of V′E to V′CO2, expressed either as a slope relating the change of V′E per unit change in V′CO2 (V′E/V′CO2 slope) or as a ratio at a fixed point in time (the ventilatory equivalent for CO2 (EqCO2)). While increased physiological dead space may be more relevant to the worse ventilatory inefficiency seen in chronic thromboembolic pulmonary hypertension (CTEPH) [25], it is likely that chronic hyperventilation, low PaCO2, is the main driver of ventilatory inefficiency in PAH [25], and indeed early computer modelling has suggested that hyperventilation may contribute to increased physiological dead space fraction [26]. Ventilatory inefficiency has also been shown to be worse in pulmonary veno-occlusive disease compared with PAH, with commensurate worsening of dyspnoea. It is not clear whether this simply relates to disease severity or a distinct difference in gas exchange phenotype, although it is plausible that there is altered ventilation–perfusion mismatch and physiological dead space [27]. Further ventilatory inefficiency may occur in the setting of a right-to-left shunt through a patent foramen ovale [28] to maintain the carbon dioxide tension in admixed pulmonary venous and shunted blood, but in this scenario there will also be significant hypoxaemia, which will further stimulate ventilation through stimulation of the peripheral chemoreceptor reflex.
In PAH, PaCO2 is reduced due to chronic hyperventilation, which is thought to relate to heart failure and increased chemosensitivity [22–24]. This increases the ventilatory demand in relation to carbon dioxide output, already increased through early anaerobiasis. This is often referred to as ventilatory inefficiency and quantified as the ratio of V′E to V′CO2, expressed either as a slope relating the change of V′E per unit change in V′CO2 (V′E/V′CO2 slope) or as a ratio at a fixed point in time (the ventilatory equivalent for CO2 (EqCO2)). While increased physiological dead space may be more relevant to the worse ventilatory inefficiency seen in chronic thromboembolic pulmonary hypertension (CTEPH) [25], it is likely that chronic hyperventilation, low PaCO2, is the main driver of ventilatory inefficiency in PAH [25], and indeed early computer modelling has suggested that hyperventilation may contribute to increased physiological dead space fraction [26]. Ventilatory inefficiency has also been shown to be worse in pulmonary veno-occlusive disease compared with PAH, with commensurate worsening of dyspnoea. It is not clear whether this simply relates to disease severity or a distinct difference in gas exchange phenotype, although it is plausible that there is altered ventilation–perfusion mismatch and physiological dead space [27]. Further ventilatory inefficiency may occur in the setting of a right-to-left shunt through a patent foramen ovale [28] to maintain the carbon dioxide tension in admixed pulmonary venous and shunted blood, but in this scenario there will also be significant hypoxaemia, which will further stimulate ventilation through stimulation of the peripheral chemoreceptor reflex.
Beyond the respiratory compensation point in the incremental exercise test (i.e. the point at which the rate of production of lactic acid cannot be buffered by bicarbonate), pH falls, resulting in direct stimulation of ventilation and thus true hyperventilation and a further fall in PaCO2. Because the buffering capacity in PAH is reduced due to the compensatory metabolic acidosis related to chronic hyperventilation, this may result in an excessive increase in ventilation during intense exercise.
While most patients are limited by a lack of cardiovascular reserve at peak exercise in PAH, as evidenced by high peak respiratory exchange ratios and lactates at the end of exercise, rather than by a lack of breathing reserve, dyspnoea remains a cardinal symptom of pulmonary hypertension, even if not fundamentally the limiting factor on an incremental test. It is not entirely clear what drives dyspnoea, with one study showing that Borg dyspnoea ratings were higher at lower absolute levels of ventilation than in healthy subjects [29]. This same study demonstrated, however, that 60% of patients with PAH had reduced expiratory flows at low lung volumes and that dynamic hyperinflation accounted for 50–60% of the variance of Borg dyspnoea ratings during cycle ergometry [29].
More often than not, cycle ergometry is used to assess the cardiopulmonary response to incremental exercise, but there are important differences in gas exchange and dyspnoea between cycling and walking, with the latter most likely to reflect everyday symptoms. Valli et al. [30] demonstrated that ventilatory inefficiency is considerably worse on walking than cycling, with higher dyspnoea at peak exercise, although absolute levels of exercise as assessed by oxygen consumption and V′CO2 were lower. They suggested that this was related to worsened ventilation–perfusion matching, but also noted a more rapid and shallower breathing pattern during walking exercise. This highlights that while cycle ergometry may offer many advantages, such as standardisation of workload, it may not provide the best representation of patients' functional limitation.
Both increased ventilatory demand and dynamic hyperinflation place increased demand on the respiratory muscles. Unlike muscles, which may increase in strength/endurance with increased loading, there is mixed evidence of respiratory muscle weakness in pulmonary hypertension [12, 29, 31, 32]. Although this does not lead to exercise limitation, it is conceivable, although unproven, that this may contribute to the sensation of dyspnoea.
Finally, there is likely to be significant interplay between psychological status and dyspnoea. Although the breathing pattern in PAH has been shown to be rapid and shallow [29], frank breathing pattern disorders, such as hyperventilation, may coexist with PAH, just as they may in other cardiorespiratory disorders. However, dyspnoea, as assessed by the Dyspnoea-12 questionnaire, shows stronger correlations with anxiety and depression scores than functional class or 6-min walking distance [5], highlighting the complex nature of dyspnoea and how it may be influenced by emotional and psychological health.
Cardiopulmonary exercise physiology in relation to disease severity in PAH
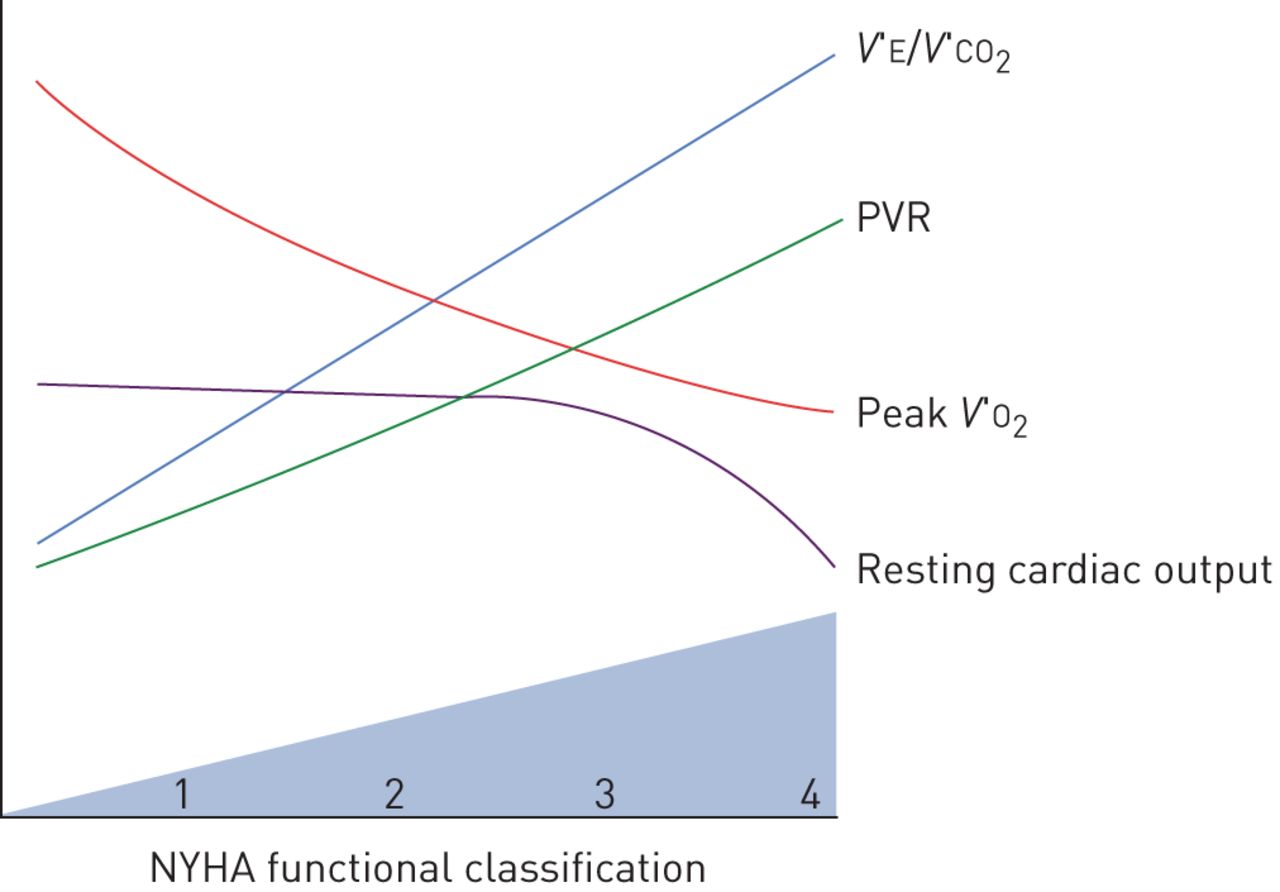
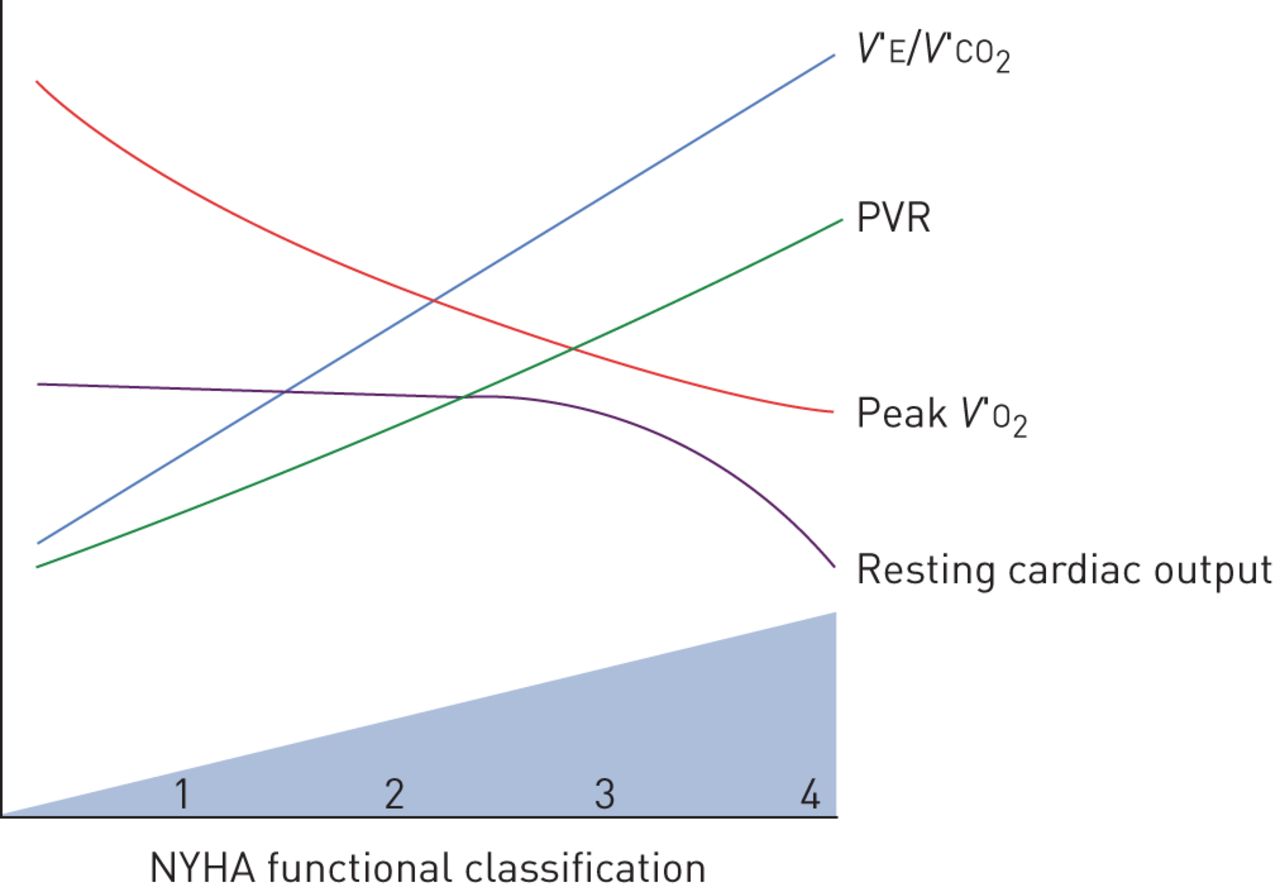
The severity of PAH can be categorised in terms of haemodynamic variables, functional capacity by New York Heart Association (NYHA) classification, or by the degree of impairment in peak V′O2 during incremental cardiopulmonary exercise testing. With progressive severity of PAH, interactions between the heart, lungs, peripheral muscle and autonomic nervous system become increasingly complex. Interestingly, no studies have specifically investigated the relationship between the haemodynamic severity of PAH and dyspnoea intensity during exercise testing. As expected, patients with worse NYHA functional class also have lower peak V′O2, higher V′E/V′CO2 slope and higher EqCO2, but the correlations between these variables and NYHA are modest, reinforcing the subjective and multifactorial nature of the NYHA classification [33]. Resting haemodynamic variables such as mean pulmonary arterial pressure, cardiac output and pulmonary vascular resistance also correlate with exercise capacity in terms of peak V′O2 or NYHA functional class (figure 3), but a substantial portion of inter-individual variability in exercise capacity cannot be explained by the haemodynamic severity [33, 34]. As such, patients with higher resting mean pulmonary arterial pressure tend to have lower peak V′O2 and higher V′E/V′CO2, but with considerable variation in ventilatory inefficiency between patients of comparable haemodynamic severity [34, 35].
{kind=link}
{kind=link}
{kind=link}
Progressive physiological changes in pulmonary arterial hypertension. V′E: minute ventilation; V′CO2: carbon dioxide production; PVR: pulmonary vascular resistance; V′O2: oxygen uptake; NYHA: New York Heart Association.
As already discussed, among the major determinants of exercise capacity in PAH are 1) the ability to increase cardiac output during exercise to meet metabolic demand, 2) the ability of the pulmonary circulation to accommodate increased pulmonary blood flow, and 3) impaired skeletal muscle function. Recently, Hasler et al. [36] studied PAH patients undergoing supine exercise by cycle ergometry during right heart catheterisation, and found that the increase in cardiac index during exercise negatively correlated with disease severity and was a strong independent predictor of survival (hazard ratio 0.25, p=0.04). Patients with mild PAH (NYHA class I and II) were able increase cardiac output significantly during exercise [36], but even mildly symptomatic patients still have reduced exercise capacity compared to healthy individuals [33]. Because pulmonary vascular resistance is still elevated in mild PAH patients, there is a disproportionate increase in mean pulmonary arterial pressure as cardiac output rises.
One mechanism by which this may limit exercise capacity in mild PAH is through ventricular interaction. Pulmonary arterial and right ventricular pressures increase substantially during exercise, which can shift the interventricular septum in diastole, impairing left ventricular diastolic filling, stroke volume and cardiac output [37]. In contrast, patients with NYHA class III or IV symptoms have lower cardiac output and lower mixed venous oxygen content at rest, and demonstrate a diminished ability to increase cardiac output during exercise [36]. Several studies of PAH patients have shown a reduced ability to augment stroke volume during exercise, which necessitates an excessive increase in heart rate to increase cardiac output [38–40]. Consequently, the oxygen pulse (V′O2/heart rate), which is an indirect estimate of stroke volume during cardiopulmonary exercise testing, is reduced in mild PAH patients in comparison to healthy individuals and is even further reduced in severe PAH patients [33].
Functional capacity and V′E/V′CO2 in PAH are influenced by autonomic nervous system hyperactivity in addition to cardiac and gas exchange impairment [41, 42]. For example, although VD/VT is elevated in patients with PAH and is associated with severity [25], high VD/VT explains only part of the increase in V′E/V′CO2 and EqCO2 [25, 35]. Therefore, mechanisms other than increased dead space, such as enhanced chemosensitivity or hyperventilation due to a reduced PaCO2 set-point, contribute to the excessive ventilatory response and high V′E/V′CO2 in severe PAH. When right ventricular function and cardiac output are impaired, low mixed venous oxygen content and hypoxaemia are exaggerated during exercise. Thus, more severe impairment in the cardiovascular response to exercise leads to increased sympathetic drive and higher ventilatory response for a given metabolic demand (i.e. higher V′E/V′CO2). This may explain why V′E/V′CO2 not only reflects the severity of PAH, but is also a powerful predictor of prognosis, as it integrates the function of the cardiac, respiratory and nervous systems [10]. While the V′E/V′CO2 slope is usually elevated in the range of 35–45 in mild PAH patients [33], a V′E/V′CO2 slope >60 (or EqCO2 >54) is typically observed in severe patients, which portends a significantly increased risk of death [43, 44]. A low end-tidal carbon dioxide tension (PETCO2) and PaCO2 are also useful markers of PAH severity during exercise testing, as they reflect inefficient ventilation due to both high dead space and altered chemoreflex sensitivity with a lower PaCO2 set-point [22, 25, 34]. Idiopathic pulmonary arterial hypertension (IPAH) patients with low resting PaCO2 (those who hyperventilate at rest) have a lower cardiac index, lower mixed venous oxygen saturation and worse survival [22]. During exercise, the excessive ventilatory response seen in PAH patients further dilutes PETCO2, causing a progressive decline in PETCO2 during exercise, which is also proportional to the severity of disease [34].
Exercise limitation phenotypes in different forms of pulmonary hypertension
Pulmonary hypertension consists of a group of complex and heterogeneous diseases and, as such, there is no homogeneous pattern of exercise limitation among different groups of the 2013 pulmonary hypertension classification from the 5th World Symposium on Pulmonary Hypertension in Nice, France [45]. Publications on characteristic mechanisms or patterns of exercise intolerance and dyspnoea for different pulmonary hypertension forms are limited; however, there is a growing number of studies dedicated to exercise limitation in pulmonary hypertension subgroups. Because this review focuses mainly on PAH, we shall only briefly discuss other forms of pulmonary hypertension in order to contrast with PAH.
Patients with combined lung disease and heart disease often have a particularly severe phenotype, characterised by severe dyspnoea and impaired gas exchange. For example, in coexistent chronic obstructive pulmonary disease (COPD) and heart failure, there is a worsening of dead space ventilation, as indicated by a raised V′E/V′CO2 slope intercept [46]. In pulmonary hypertension, however, the impact of coexisting lung disease is perhaps best illustrated in a detailed physiological study using invasive exercise haemodynamics in patients with COPD and different levels of severity of pulmonary hypertension [47]. Patients with mild and moderate pulmonary hypertension (i.e. somewhat more in proportion to their COPD severity) displayed ventilatory limitation with relatively preserved V′E/V′CO2 relationships and hypercapnia at peak exercise indicative of respiratory mechanical limitation; however, patients with severe pulmonary hypertension behaved more like those with PAH, showing high V′E/V′CO2 slopes, hypocapnia and an exhausted circulatory reserve. Thus, despite a “double hit” of lung disease and severe pulmonary hypertension, this group of severe patients may be those whose symptoms of dyspnoea benefit from PAH therapies, although this has yet to be proven in randomised controlled studies. This study also serves to demonstrate that when PAH and mild coincidental lung disease coexist, such as may occur in scleroderma, the circulatory changes of PAH tend to dominate symptoms and exercise limitation.
CTEPH shares many similarities with PAH, but there are reasons to believe that they may differ in terms of the mechanisms of dyspnoea, in particular illustrated by the commonly encountered situation of chronic thromboembolic disease in the absence of pulmonary hypertension, which often results in significant symptoms of dyspnoea. Two studies have compared exercise gas exchange patterns in CTEPH and PAH [25, 48]. Both studies conclude that significant differences in gas exchange patterns exist, most probably due to a higher physiological VD/VT in CTEPH compared to PAH, due to vascular obstruction by organised thrombosis. This may lead to a dissociation between pulmonary hypertension severity (in terms of the impact on right ventricular function) and gas exchange in CTEPH, unlike PAH [49], such that dyspnoea may be less reflective of prognosis in CTEPH than PAH [25].
Impact of comorbidities on exercise limitation/breathlessness in PAH
PAH often presents as an isolated condition, particularly in “classic” IPAH in young individuals, but may coexist with other conditions. A fine line exists between IPAH with coexisting lung disease, such as mild incidental emphysema or asthma, where IPAH would have presented whether the incidental lung disease had been present or not, and group 3 pulmonary hypertension, where pulmonary hypertension is considered secondary to lung disease and would not have existed without it. In this review, we consider the former scenario, where it would be anticipated that the interaction of minor parenchymal lung disease and IPAH would lead to worsened ventilatory inefficiency and hypoxaemia on exercise, leading to worsened dyspnoea relative to cardiovascular symptoms when compared with isolated IPAH. Furthermore, this is exacerbated where this is coupled with reduced lung volumes. Few published data exist, but this is a cardiopulmonary pathophysiological phenotype that we recognise in our laboratories, and is similar to that seen in severe pulmonary hypertension “out of proportion” to COPD [47].
Obesity places an additional burden on cardiopulmonary function [50]. There is an oxygen cost of unloaded exercise, largely due to the increased/hidden work of moving increased limb weight against no resistance [51]. In addition, it has been noted that otherwise healthy patients with obesity have a higher oxygen cost of breathing, which may relate to the increase in the work of breathing through altered respiratory mechanics, such as increased intrathoracic pressure and changes in airway resistance due to reduced functional residual capacity [50]. This may incur a small, yet relevant, oxygen steal during exercise. Although the increased work of breathing may not be clinically relevant in healthy obese subjects in producing worse dyspnoea, there is increased ventilatory demand in PAH, and we can thus speculate that this may become more important.
Iron deficiency has recently been recognised as an important comorbidity of IPAH and is associated with worse prognosis and exercise capacity. It has been suggested that iron deficiency is not just a marker of severity, but independently contributes to poor prognosis and exercise function [52]. No studies have assessed the impact of iron replacement on prognosis, but two open-label protocols have demonstrated improved aerobic capacity following intravenous iron, most probably due to improved skeletal muscle mitochondrial oxidative capacity [53, 54]. Data were not reported on dyspnoea specifically, but improvements were seen in both studies in quality of life as assessed by the SF-36 score (36-item Short-Form Health Survey).
It is debatable whether skeletal muscle, including both the peripheral locomotor groups and respiratory muscles, is considered a comorbidity of PAH or an integral part of the “PAH syndrome”. In IPAH, as well as other forms of pulmonary hypertension isolated to the cardiopulmonary system, such as CTEPH, there are many studies documenting impaired skeletal muscle function. Clearly, in other forms of PAH that are part of a more formal systemic syndrome such as the connective tissue diseases, myopathy may be more severe, sometimes requiring specific therapy. In these cases, dissociating primary cardiopulmonary limitation from muscle disease is important, not only to make the appropriate diagnosis, but also to maximise the opportunity to treat muscle disease and to avoid overtreatment of pulmonary hypertension. Primary muscle disease may often present with muscle pain on exercise, but can also contribute to dyspnoea through early anaerobiasis, thus increasing ventilatory demand.
Limitations
Although exercise dyspnoea is a well-known and cardinal symptom of PAH, its complex multifactorial origin makes it difficult to understand and relate back to measurable pathophysiological mechanisms. The best measurement tools we have derive from exercise testing, but dyspnoea scores and questionnaires only provide us with crude mechanistic insight. Objective measurement of dyspnoea potentially requires more sophisticated instruments such as functional neurological imaging. Therapies targeting dyspnoea directly are frequently used at the end of life in palliative care settings, but none has been studied in less advanced disease, and while it may be feasible to do this, without better understanding the mechanisms and pathophysiological surrogates of dyspnoea, these therapeutic areas will be hard to explore.
Conclusion
Dyspnoea is the most common and often most debilitating symptom of PAH, and while the physiology of PAH is well characterised, dyspnoea itself remains elusively misunderstood. Treatments that target PAH directly lead to improvements in dyspnoea, but it remains possible that opportunities exist to target dyspnoea directly without having an impact on the underlying disease process. Unless dyspnoea is better understood, these opportunities are likely to remain underexplored.
Disclosures
Supplementary Material
O. Sitbon ERR-0039-2017_Sitbon
Footnotes
Number 6 in the Series “Exertional dyspnoea” Edited by Pierantonio Laveneziana and Piergiuseppe Agostoni
Previous articles in this series: No. 1: Dubé B-P, Agostoni P, Laveneziana P. Exertional dyspnoea in chronic heart failure: the role of the lung and respiratory mechanical factors. Eur Respir Rev 2016; 25: 317–332. No. 2: O'Donnell DE, Elbehairy AF, Faisal A, et al. Exertional dyspnoea in COPD: the clinical utility of cardiopulmonary exercise testing. Eur Respir Rev 2016; 25: 333–347. No. 3: Bernhardt V, Babb TG. Exertional dyspnoea in obesity. Eur Respir Rev 2016; 25: 487–495. No. 4: Bonini M, Fiorenzano G. Exertional dyspnoea in interstitial lung diseases: the clinical utility of cardiopulmonary exercise testing. Eur Respir Rev 2017; 26: 160099. No. 5: Weatherald J, Lougheed MD, Taillé C, et al. Mechanisms, measurement and management of exertional dyspnoea in asthma. Eur Respir Rev 2017; 26: 170015.
Support statement: Jason Weatherald is the recipient of a joint European Respiratory Society/Canadian Thoracic Society Long-Term Research Fellowship (LTRF 2015-4780).
Conflict of interest: Disclosures can be found alongside this article at err.ersjournals.com
Provenance: Commissioned article, peer reviewed.
- Received April 1, 2017.
- Accepted June 3, 2017.
- Copyright ©ERS 2017.
ERR articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References











Jump To
- Article
- Abstract
- Abstract
- Introduction
- Cardiovascular response to exercise in PAH
- Respiratory response to exercise in PAH
- Cardiopulmonary exercise physiology in relation to disease severity in PAH
- Exercise limitation phenotypes in different forms of pulmonary hypertension
- Impact of comorbidities on exercise limitation/breathlessness in PAH
- Limitations
- Conclusion
- Disclosures
- Footnotes
- References
- Figures & Data
- Info & Metrics