





Abstract
The pathological and molecular classification of lung cancer has become substantially more complex over the past decade. For diagnostic purposes on small samples, additional stains are frequently required to distinguish between squamous cell carcinoma and adenocarcinoma. Subsequently, for advanced nonsquamous cell nonsmall cell lung carcinoma (NSCLC) patients, predictive analyses on epidermal growth factor receptor, anaplastic lymphoma kinase and ROS1 are required. In NSCLCs negative for these biomarkers, programmed death ligand-1 immunohistochemistry is performed. Small samples (biopsy and cytology) require “tissue” management, which is best achieved by the interaction of all physicians involved.
Abstract
Tissue management is required to perform diagnostic and predictive testing in nonsmall cell lung carcinoma http://ow.ly/Ogeq30adDvN
Introduction
The pathological and molecular classification of lung cancer has become substantially more complex over the past decade. Until the arrival of pemetrexed, a relatively simple distinction between small cell lung cancer (SCLC) and nonsmall cell lung cancer (NSCLC) was sufficient to guide systemic treatment, and a meta-analysis of four different platinum doublets showed that none offered a significant advantage over the others in the treatment of advanced NSCLC [1]. However, since then, multiple phase III trials showed that histology directs treatment and guides molecular profiling. Selected agents (e.g. pemetrexed and nintedanib) show superior efficacy in adenocarcinomas and large cell carcinomas when compared to squamous cell carcinomas (SqCC) [2–5]. Molecular alterations that drive tumorigenesis (driver mutations) are predominantly found in NSCLC, and the European Society for Medical Oncology guideline indicates that molecular testing is only recommended in these tumours and not in patients with SqCC, except for never-/former light smokers (<15 pack-years) [6].
In patients with sensitising epidermal growth factor receptor (EGFR) mutation, anaplastic lymphoma kinase (ALK) or ROS1 rearrangement, treatment with oral tyrosine kinase inhibitors (TKIs) have been approved by the European Medicines Agency (EMA) and US Food and Drug Administration (FDA) as the preferred choice of treatment.
The ALK inhibitors crizotinib and ceritinib are superior to standard chemotherapy in ALK-positive NSCLCs, supporting ALK testing for advanced-stage NSCLC [7, 8]. Therefore, all patients with advanced nonsquamous NSCLC should be evaluated for the presence of these molecular alterations. In addition, multiple other driver genes have been identified (e.g. HER2, BRAF, RET, MET and NTRK1) and targeted drugs are available or being developed for these alterations.
As a result, the pathological diagnosis of lung cancer has become a multistep process, beginning with morphology and immunohistochemistry (IHC) to discriminate benign from malignant process, metastases from primary lung cancer and small cell lung carcinoma and NSCLC. In cases of primary nonsquamous NSCLC, this process is followed by molecular characterisation.
Re-biopsy upon disease progression has long been a mainly scientific effort, but with osimertinib, the first registered drug for EGFR TKI resistance (i.e. T790M mutation) [9], re-biopsy should be the standard of care in selected cases. Besides the secondary EGFR mutation, other resistance mechanisms such as HER2 and MET amplification have been described [10], and drugs for these bypass resistance mechanisms are currently under investigation in clinical trials. In cases of ALK translocation, specific ALK resistance mutations develop after ALK TKI and the type of mutation correlates with the sensitivity to next generation ALK TKIs [11]. Thus, depending on the clinical context, specific molecular tests need to be requested for re-biopsies. Liquid biopsies (i.e. analysis of cell-free tumour DNA from blood plasma) have become another important option in such a situation, but are not within the scope of this review. Ideally, pulmonologists and oncologists require diagnoses that are rapid, precise and relevant for therapeutic management. The possibilities of the small samples to meet these needs are limited; a realistic view of these possibilities is presented here.
While the number of tests over the past decade has increased, sample size (predominantly small biopsies or cytological specimens) has decreased. A variety of techniques are available for tissue analysis and tissue characterisation (amount and tumour percentage). Next-generation sequencing (NGS) can potentially test for all molecular alterations at a low level or with <10% of tumour cells, but a minimum amount of tissue is still needed, allowing the extraction of 10–150 ng DNA or RNA, depending on whether PCR- or capture-based NGS technology is used. In addition, sequential testing is still common in many laboratories, with incomplete molecular profiling in cases with insufficient tumour amount or quality.
Therefore, it is critical that the requesting physician is aware of the techniques that are applied in the lab and what tissue characteristics are required for the requested parameters. The pathologist should preserve precious tissue for specific (molecular) analyses; the pathologist needs to be a full member of the multidisciplinary team, being informed about the clinical presentation, and obtaining an informative request form and feedback of treatment results in selected cases (e.g. rare molecular alterations).
In this article, we review the pathological analysis of NSCLC on small biopsies and cytology specimens, emphasising on the clinical perspective.
Pathology diagnosis
The previous 2004 World Health Organization (WHO) classification of lung cancer was based on resection specimens [12]. In the recent 2015 WHO classification, recent literature-specific issues with small biopsy and cytology samples are addressed [13, 14]. In biopsies, the distinction between adenocarcinoma and SqCC made using the routine haematoxylin and eosin (H&E) stain is not possible in up to 30–40% of NSCLC cases, because of the small sample size (∼1 mm) and the reduced chance of sampling a differentiated part of the tumour [15]. For this situation the diagnosis of NSCLC not otherwise specified (NOS) is used, which leads to the application of a panel of three IHC markers, thyroid transcription factor (TTF)1 and mucin stain for glandular lineage and p63/p40 for squamous cell lineage [16]. The threshold for positivity is different for TTF1 and p63/p40. TTF1 is considered positive, if weak but certain staining is present [15]. However, in most TTF1-positive lung carcinomas the staining is +++ in the majority of the tumour cell nuclei. p63/p40 is strongly (+++) positive in almost all nuclei of undifferentiated neoplastic cells in SqCC, especially in the periphery of a field of tumour cells, with loss of staining in more differentiated areas. The threshold for p63 IHC determining SqCC lineage is high; the majority (>80%) of the nuclei need to be strongly positive [15, 17, 18]. However, currently p40 is preferred over p63. p40+, TTF1−, mucin− tumours without diagnostic features of keratinisation or intercellular bridges should be diagnosed as “p40/p63-positive NSCLC-NOS, consistent with non-keratinising SqCC”. For p40/p63−, TTF1+ and/or mucin+ tumours the diagnosis is “TTF1+ (or mucin+) NSCLC-NOS, consistent with solid type adenocarcinoma”. p63, and to a much lesser extent p40 may show weak (+ to ++) and heterogeneous staining in adenocarcinoma, which should not mislead investigators to a diagnosis of SqCC. Rare tumours positive for both lineage-determining markers may represent adenosquamous carcinomas or peripheral adenocarcinomas with squamous differentiation [19–21]. Another small fraction of tumours (<10%) are negative for both lineages and qualify as NSCC-NOS. With this approach, ∼85–90% of the biopsies that cannot be subtyped by morphology alone are segregated to restrict predictive mutation analysis to adenocarcinomas and NSCLC-NOS. Liberal use of lineage (TTF1 and p40/p63) is advocated, as the H&E staining may be misleading, and some SqCC may mimic adenocarcinoma and vice versa[15, 22]. An erroneous diagnosis of SqCC may prohibit predictive analysis. Note that the diagnosis of “large cell carcinoma” is only made on resection specimens when additional stains are not informative (“large cell carcinoma null phenotype”). Overall, this diagnostic approach provides biological correlation as well as predictive relevance for chemotherapy [2, 3, 5].
Predictive analysis
General considerations
The implication of predictive testing is that the test outcome is essential for decisions about drug treatment. Although in theory this may also incorporate the effect of xenobiotic metabolism and dosage of the drug, in this review only NSCLC tumour testing for choice of drug is discussed.
The algorithm including NGS testing in clinical practice is shown in figure 1.

Testing algorithm integrating diagnostic and predictive analysis in nonsmall cell lung cancer (NSCLC). Note the financial implication: the costs for predictive testing are added to those for diagnostic testing. FFPE: formalin-fixed paraffin-embedded; IHC: immunohistochemical; TTF: thyroid transcription factor; EGFR: epidermal growth factor receptor; HER: human epidermal growth factor receptor; ALK: anaplastic lymphoma kinase; NGS: next-generation sequencing; PD-L: programmed death ligand; FISH: fluorescence in situ hybridisation; NTRK: neurotrophic tropomyosin receptor kinase.
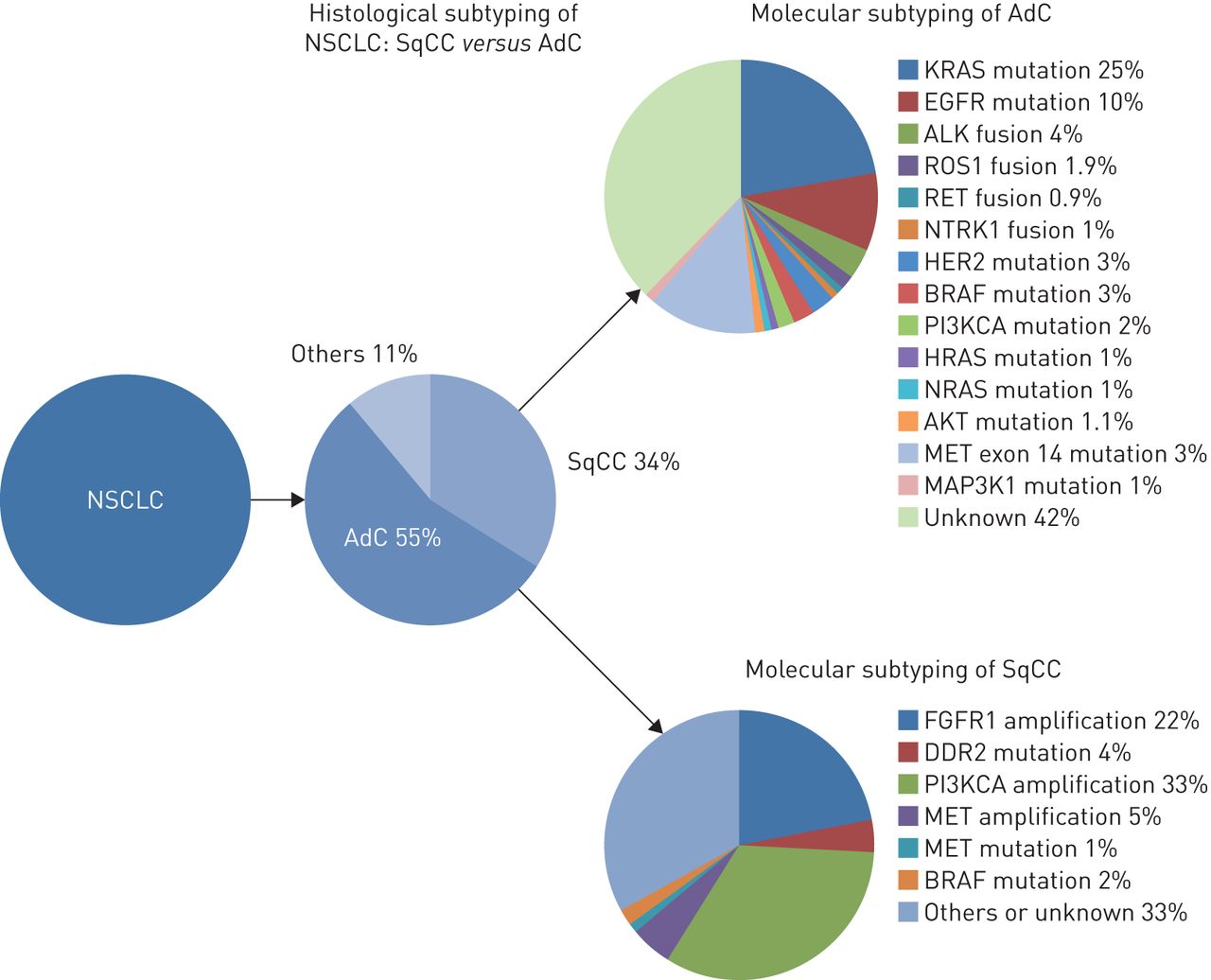
The genetic abnormalities in NSCLC include EGFR (exon 21 L858R mutation and exon 19 deletion) mutations, occurring in 11% of Caucasian patients and up to 60% of Asian patients; HER2 mutations (1%); BRAF mutations (2%); exon 14 MET mutations (3.3%); and ALK, ROS1, RET and NTRK rearrangements reported in 5%, 2%, 1.9% and 0.9%, respectively (figure 2). Gefitinib, erlotinib and, more recently, afatinib, have been approved for the first-line treatment of EGFR-mutated advanced NSCLC, and the ALK-inhibitors crizotinib and ceritinib are superior to standard chemotherapy in ALK-positive NSCLCs. In addition, crizotinib has been approved by the FDA for ROS1 rearrangement-positive NSCLC patients, and new targeted therapies gave encouraging results in early-phase clinical trials for NSCLC patients with BRAF [23, 24], HER2 [25] or exon 14 MET mutations [26, 27], and RET [28, 29] or NTRK [30, 31] rearrangements.

Histological and molecular subtypes in nonsmall cell lung carcinoma (NSCLC). SqCC: squamous cell carcinoma; AdC: adenocarcinoma.
Most of these genetic alterations occur in nonsmokers, and ALK and ROS1 rearrangements arise predominantly in young patients and in adenocarcinomas with solid or cribriform patterns and/or signet ring cells. In contrast, KRAS mutations are the most frequent (nearly 30% of NSCLCs) and predominate in smokers, but they are not targetable to date [32]. In this new era of personalised medicine, which also includes immune checkpoint inhibitors, pathologists play a crucial role in the characterisation of tumour samples and determining whether they are appropriate for molecular assays. In addition, despite the development of NGS, pathologists are still involved in chromosomal rearrangement detection by IHC and fluorescence in situ hybridisation (FISH), and they will be increasingly asked to assess programmed death ligand (PD-L)1 expression to select patients for treatment with PD1/PD-L1 inhibitors. However, as small biopsies and cytology specimens are the only material available for almost 80% of NSCLC patients, pathologists need to make most efficient use of the tumour specimens and prioritise the analyses.
Formalin fixation and paraffin embedding, which is a global standard to preserve tissue morphology for diagnosis, modifies nucleotides (C to T and G to A changes). Formalin provokes cross-linkings between proteins and nucleic acids, which render them inaccessible to enzymes used in molecular assays [33, 34]. Highly acidic solutions used for decalcification of bone metastases represent another challenge since they degrade the tumour DNA. Here, the simultaneous collection of material using fine needle aspiration (FNA) for ethanol-fixed slides and/or cell blocks is recommended, as it accelerates the diagnostic process and enables predictive marker analyses without the limitations of decalcification. Accordingly, the importance of careful fixation and processing of tumour samples for molecular analysis has been emphasised in recent guidelines [35, 36].
Mutation detection
NSCLC tissues are heterogeneous and composed of a mix of “normal” stroma cells (immune cells, fibroblasts and endothelial cells), malignant cells and “normal” cells from surrounding tissues. Thus, the molecular assay implemented in each laboratory must be sensitive enough to overcome the dilution of tumour DNA with DNA from normal cells. For instance, a limit of detection of 10% corresponds to the presence of a heterozygous mutation (mutated allele) in 20% of malignant cells. As an example, in 2012, among the 20 000 NSCLC samples analysed for EGFR mutations on the 28 INCa (French National Cancer Institute) genetics platforms, 5.2% did not contain enough tumour cells (<10%) to enable the detection of mutations [32, 37].
Pathologists are involved in the assessment of the tumour cell percentage, but a high interobserver variation exists between them [38], probably due to inadequate definition of cellularity, and discordance in estimating the percentage of tumour cells and the percentage of tumour area. Fortunately, the interobserver variations predominate for specimens with a low percentage of tumour cells [39]. In a study by Molina-Vila et al. [40], 81% of samples containing <150 tumour cells, considered initially as nonavailable for mutations analyses, were available after laser capture microdissection. To date, many laboratories routinely enrich samples in tumour cells by using macro- or microdissection. The sandwich H&E procedure (H&E analysis of the sections before and after those used for DNA/RNA analyses) aims to guarantee the presence of tumour cells in the sections used for molecular analysis. It is very important to emphasise that not only the amount and percentage of tumour cells is important, but also the amount of total tissue and its number of cells itself; as an example, for most PCR-based NGS techniques, a minimum of 5–10 ng of total DNA (including malignant and normal DNA) is required, equal to 1000 cells in formalin-fixed and paraffin-embedded tissue material [41]. In contrast, hybrid capture-based NGS technologies, encompassing RNA sequencing, require 100–200 ng of RNA or DNA. In this latter case, up to one-third of all specimens may be insufficient for NGS analysis [42].
Mutations can be detected using conventional Sanger sequencing, amplification refractory mutation assays, restriction fragment length polymorphisms and by targeted NGS panels (table 1). Bidirectional Sanger sequencing without an enrichment step has a limit of detection of 10–25% of total DNA, leading to some false negative results, while PCR-based ultrasensitive and NGS methods using gene panels can detect mutation in samples containing <10% of tumour cells, but can generate artefacts leading to false positive results, i.e. artificial base pair changes that may erroneously be taken for mutations. Whole exome sequencing and whole genome sequencing analyses require greater amounts of DNA (in the order of micrograms from close to 106 tumour cells for whole genome sequencing) and are not adapted for small samples.
Molecular techniques used for predictive testing, ranked for different categories of sensitivity
For small biopsies and cytology specimens containing sufficient tumour cells, the success rate for EGFR genotyping ranged according to series from 83% to 100% [44–46]. The anatomic site and the procedure used to obtain the biopsy/cytology specimen could influence the yield of the testing, particularly when an “on site” procedure with touch imprints is performed, reducing the number of tumour cells remaining on the specimen, or when bone metastases requiring decalcification are considered [47].
ALK rearrangement
ALK gene rearrangements lead to the expression of ALK fusion proteins with strong oncogenic properties [32, 48]. Numerous algorithms and guidelines have been proposed for the detection of ALK rearrangements. Initially, FISH was the gold standard. However, FISH is a technically demanding method, requiring specialised equipment and an experienced technician/pathologist. For this reason, ALK IHC has become a widely used, cost-effective technique for prescreening and diagnosis. IHC requires limited equipment, is available in pathology laboratories worldwide and requires only small number of tumour cells to detect the presence of the fusion protein (diffuse cytoplasmic staining). In the USA the FDA recently approved the Ventana ALK (D5F3) CDx Assay (Ventana Medical Systems, Tucson, AZ, USA) as a companion diagnostic for crizotinib [49, 50]. In Europe the EMA defined patients with ALK-positive metastasised NSCLC eligible for treatment with ALK inhibitor, stimulating the development of IHC as screening method for ALK-positive NSCLC. Currently, two validated antibodies exist for ALK IHC in lung cancer: D5F3 (Cell Signaling Technology, Danvers, MA, USA) [51] and 5A4 (Leica Microsystems, Milton Keynes, UK) [52], which appear to be equally sensitive [53].
ALK IHC interpretation in tumour cells is quite straightforward, as ALK protein is not expressed in normal epithelial lung tissue (figure 3). However, pathologists should be familiar with staining in other cell types: light cytoplasmic stippling in alveolar macrophages, cells of neural origin (nerve and ganglion cells (a cross-section of appendix makes a good external control)), glandular epithelial staining, extracellular mucin and necrotic tumour areas. Neuroendocrine tumours (Merkel cell tumours of the skin or large cell neuroendocrine carcinoma) have also been associated with positive reactions [54, 55]. False-negative ALK FISH results are mainly related to paucity of tumour cells (<50 cells), the presence of hyperplastic (reactive) normal cells considered as malignant, impaired tissue preservation and fixation variations and rare complex rearrangements proving negative by FISH but positive by IHC. In contrast, false positivity of FISH is often due to atypical patterns, such as multiple fusion signals and solitary green signals with split 5′ centromeric probe. False-positive cytoplasmic staining in NSCLC has been noted with the tyramide amplification system using D5F3 [56]. The fraction of ALK IHC-positive cases that are ALK FISH-negative is low (0.4% of all tested NSCLC cases; 7% of ALK IHC-positive NSCLC), showing that ALK IHC is a useful screening method [51, 52, 57–76]. The information about response rate on ALK inhibitor of IHC-positive/FISH-negative cases is limited, but some responses have been reported [67, 77]. Of note, most discrepancies between FISH and IHC seem to occur when the percentage of rearranged cells is in a grey zone between 10% and 20%. Overall, the sensitivity and the specificity of IHC versus FISH both range from 81% to 100% [66, 71, 73, 76, 78, 79]. To maintain the reliability of the ALK IHC assay for detecting ALK positivity, laboratories should participate regularly in external quality assessment programmes. The upcoming guideline from the College of American Pathologists/International Association for the Study of Lung Cancer/Association for Molecular Pathology will probably state that positive ALK IHC is sufficient to treat patients with ALK inhibitor.

Transthoracic needle biopsy of lung adenocarcinoma: all tumour cells are stained with 5A4 clone (anaplastic lymphoma kinase (ALK) immunohistochemistry (IHC)) original magnification a) ×40 and b) ×200 (Ventana, Tucson, AZ, USA); c) split signals are observed in >15% of the tumour cells by fluorescence in situ hybridisation (FISH) using break-apart probe. d) Biopsy of another patient with adenocarcinoma, where nearly all tumour cells are all stained by D4D6 clone (ROS1 IHC Ventana; original magnification ×200); e) split signals are observed in >15% of the tumour cells by FISH using break-apart probe (ZytoVision, Bremerhaven, Germany).
ROS1 rearrangement
Data on the predictive value of ROS1 IHC are still evolving. Most studies on ROS1 IHC use the D4D6 rabbit monoclonal antibody (figure 3) (Cell Signaling Technology) [80]. Although ROS1-rearranged tumours are invariably and diffusely positive with D4D6 antibody (cytoplasmic staining), ROS1 IHC positivity has been reported in some EGFR- or Her2- mutated or RET-rearranged tumours or in mucinous adenocarcinoma without ROS1 rearrangement; in addition, ROS1 is expressed in normal reactive pneumocytes and macrophages [81]. In most cases, the expression in these cells is weak to moderate (+/++) in intensity [50, 82]. In contrast to ALK, where the ganglion cells of the appendix serve as an adequate external control, there is currently no external benign tissue control for ROS1 [50, 82]. Tumour specimens with known ROS1-rearrangement, or a cellblock of the HCC78 cell line harbouring the SLC34A2-ROS1 fusion gene can serve as external positive controls [82, 83].
NSCLC studies on ROS1 IHC mention variation in staining intensity with D4D6 ranging from weak (+/++) to strong (+++) [81, 84–91]. Strongly stained cases were confirmed to be ROS1-rearranged using FISH in 88% (summarised 49 out of 56 ROS1 +++ cases), while in the weakly positive cases rearrangement could only be demonstrated in 14% (summarised 31 out of 228 ROS1 +/++ cases) [81, 84–91]. Therefore, in practice, the current information suggests screening with ROS1 IHC and subsequent confirmation of the IHC-positive cases using FISH [92]. ROS1 inhibitors should only be given to patients whose tumours are double positive according to IHC and FISH. There is currently not enough information on the ROS1 FISH-positive cases with negative ROS1 IHC.
PD-L1
PD-L1 expression by IHC is a biomarker with predictive value for response to PD-(L)1 monoclonal antibodies. The pharmaceutical action is based on the inhibition of PD1 or PD-L1 and tackled by five different agents, each with its own PD-L1 antibody: nivolumab with 28-8 rabbit antibody [93]; pembrolizumab with 22C3 mouse antibody [94]; atezolizumab with SP142 rabbit antibody; durvalumab with SP263 rabbit antibody [95]; and avelumab with rabbit antibody 73-10 [96]. The commercial complementary PD-L1 diagnostic test for nivolumab (28-8 pharmDx; Dako Agilent, Santa Clara, CA, USA) and companion test for pembrolizumab (22C3 pharmDx; Dako) are now FDA-approved for use in NSCLC. The different commercial assays are currently being compared to each other and there are attempts to calibrate laboratory developed tests to the clinical validated assays. Together with growing data on the variation in read-outs, this might eventually lead to a test equivalence applicable for different drugs in the near future.
The tissue handling procedure for PD-L1 IHC does not principally differ from other diagnostic or predictive IHC markers such as ALK [97]. Critical analytical steps are minimum fixation time of 3–6 h; cut unstained slides, preferably not older than 3–6 months and specimen age <3 years [98]. The use of PD-L1 IHC on decalcified tissues should be interpreted with caution until further validation studies on PD-L1 IHC have become available.
PD-L1 expression may be present on dendritic cells, macrophages, mast cells, T- and B-lymphocytes, endothelial cells and tumour cells [99]. Any background staining should be of less than + staining intensity. Currently, not all definitions for PD-L1 positivity are the same for the five potential assays. In four commercial assays, PD-L1 staining is defined as complete circumferential or partial linear plasma membrane staining of tumour cells at any intensity. Only cytoplasmic staining in tumour cells is not considered positive for scoring purposes. In the fifth assay (SP142), the presence of PD-L1-positive immune cells is also used to call the PD-L1 staining positive. To a certain extent, this may hamper the attempt to establish a single PD-L1 IHC test equivalent to all clinical validated tests, and possibly lead to two tests for broader application. In PD-L1 the immune activation response at the tumour–stroma interface may also play a role, not only in the interpretation, but also a possible effect of (not) sampling: smaller tumour biopsies may miss the pertinent tumour–immune interface and therefore generate lower PD-L1 IHC scores than if the tumour–stroma interface was sampled. One study reported a 92% concordance between biopsy and resection [100], while another reported a much lower (52%) concordance rate with underestimations in the biopsies [101]. Possibly, sampling variation in relation to heterogeneity in PD-L1 expression may explain the ∼9% of patients who react on PD-1/PD-L1 treatment with negative PD-L1 IHC staining [102, 103]. Pitfalls towards false-positive results may be PD-L1 positive histiocytes laying against PD-L1 negative tumour cells and cytoplasmic staining only of tumour cells. For PD-L1 reporting of the predictive IHC test, we recommend reporting the percentage of tumour cells as well as immune cells, so that the clinicians can align the applicable thresholds to the different available drugs. In cases of absent PD-L1 staining in NSCLC, the result of the positive controls should also be reported. As PD-L1 IHC is currently only validated on histological material and is a biomarker used to direct first-line treatment, histology is preferred over cytology.
Cytology
The necessity of performing predictive biomarker analyses in NSCLC specimens has drawn attention to cytology, since up to 40% of all advanced NSCLC are diagnosed by cytology alone [14, 16, 104]. Cytology is not only equivalent to histology for subtyping NSCLC, but also an attractive, minimally invasive method to collect tumour material for repetitive biomarker analysis on recurrent or metastatic disease [105]. Endoscopic ultrasound guided transbronchial fine-needle aspiration is among the most rewarding cytologic techniques. However, other modalities, such as transthoracic FNA, bronchial secretions or brushes, bronchoalveolar lavage and pleural effusions or FNA from distant metastatic sites are also important. FNAs are typically smeared or centrifuged onto glass slides, fixed using alcohol-based solutions or sprays and stained with Papanicolaou or Diff-Quik stain. Liquid-based cytology (LBC) is an alternative format by which the needle-rinse is collected in an alcohol-based fixative for semi-automated preparation of a monolayer slide by sedimentation or filter techniques. Finally, the residual material from FNAs or LBCs can be processed to form formalin-fixed paraffin-embedded (FFPE) cell blocks [106]. Cell blocks have become the preferred method for ancillary testing in many laboratories, as they can be handled in the same way as histological specimens, and the same protocols for biomarker analysis are applicable. Aside from the ability to cut multiple repeat sections, cell blocks also have the advantage of long-term preservation of protein or DNA quality. However, cell blocks are not always available, may be paucicellular or inadequate for molecular testing, and the diagnostic morphological details are inferior to those of smears/cytospins/LBC. As opposed to ethanol fixed smears/cytospins/LBC specimens, the DNA quality in cell-block specimens is impaired by formalin, leading to cross-linking and chemical modification of nucleotides.
There is much more pre-analytical variability in cytology specimens than in histological specimens [14, 49]. For instance, there are different types of fixatives with varying proportions of ethanol and the addition of various other components used for smears/cytospin preparations, LBC fixatives (e.g. ThinPrep (Hologic, Marlborough, MA, USA) or SurePath (BD Diagnostics, Oxford, UK)), and different stainings such as Papanicolaou, Diff-Quik, H&E or May–Grünwald–Giemsa. In cases of cell blocks, there is variability of the preservative solutions used prior to formalin fixation and paraffin embedding. Which of these variables are applied in a particular laboratory is usually a matter of preference, tradition and economics. All can provide equally satisfactory diagnostic results. The challenge comes with the performance of ancillary testing that may be affected by these variables in an unpredictable manner [14, 107]. This calls for rigorous validation and protocol optimisation for each individual assay, as well as continuous quality control when such testing is performed in cytological specimens.
In general, there are three main technologies for ancillary studies, including IHC, also referred to as immunocytochemistry in case of cytological specimens, FISH and mutation analysis. Ancillary testing including IHC is most commonly applied to cell blocks using the same protocols as for FFPE tissue specimens. However, IHC is also widely used on ethanol fixed smears/cytospins/LBC despite a lack of consensus on technical platforms or protocols [108]. As emphasised earlier, rigorous validation, protocol optimisation and quality control are essential when applying IHC to ethanol-fixed cytological specimens, since the performance of protocols and antibodies often differ from those used for FFPE material [109]. Notably, UK NEQAS has an external quality assessment programme in place to help standardise and improve the quality of IHC in cytology [107]. Cell lines and surplus material of clinical specimens, such as malignant effusions or touch preparations from surgical specimens can serve as appropriate positive control slides for smears/cytospins/LBC samples for the commonly used IHC markers. Cover-slipping of stained slides with mounting media protects the epitopes from degradation, allowing IHC on archival specimens that have been stored for several months at room temperature [110]. Subtyping of NSCLC using IHC in cytology has become a diagnostic standard in many laboratories [111]. In contrast, predictive marker testing in cytology by IHC is more challenging, as it is connected to assays and scoring guidelines that have been established on histological material and validated in clinical trials [104]. Predictive IHC markers that are most commonly used in NSCLC at the time of writing include ALK, ROS1 and PD-L1 (figure 1). FDA-approved assays exist for ALK and PD-L1 IHC in histology, but there are none yet for ROS1 or for cytological material. Notably, the EMA prescribes a robust and well-validated methodology for predictive ALK and ROS1 testing, but does not restrict it to a particular technique or histology. Although more data are needed, there is accumulating evidence that ALK testing using IHC can be reliably applied to both smears/cytospins/LBCs or cell blocks [110, 112]. It is foreseeable that similar data will soon become available for ROS1 IHC [50, 82]. PD-L1 IHC in cytology may pose a particular challenge given the different existing antibodies and scoring algorithms. PD-L1 staining and estimation of the percentage of positive tumour cells should be feasible in principle both on smears/cytospins/LBC and cell blocks. In smears/cytospins/LBC, membranous staining is less distinct than in FFPE tissue/cell block sections, since the cell membranes are not cut, but intact. Thus, staining of the horizontal cell membrane mostly appears as a diffuse surface staining (figure 4). Although peer-reviewed published literature is not yet available at the time of this writing, emerging data from cell blocks and matched histological specimens suggest that cytological material is as good as histological material for PD-L1 IHC tumour cell analysis [113]. Nevertheless, quantitation of PD-L1 positive immune cells will be definitely more challenging. Future studies correlating cytology-based PD-L1 IHC to treatment result (percentage positivity, not just negative or positive) are needed for clinical validation.
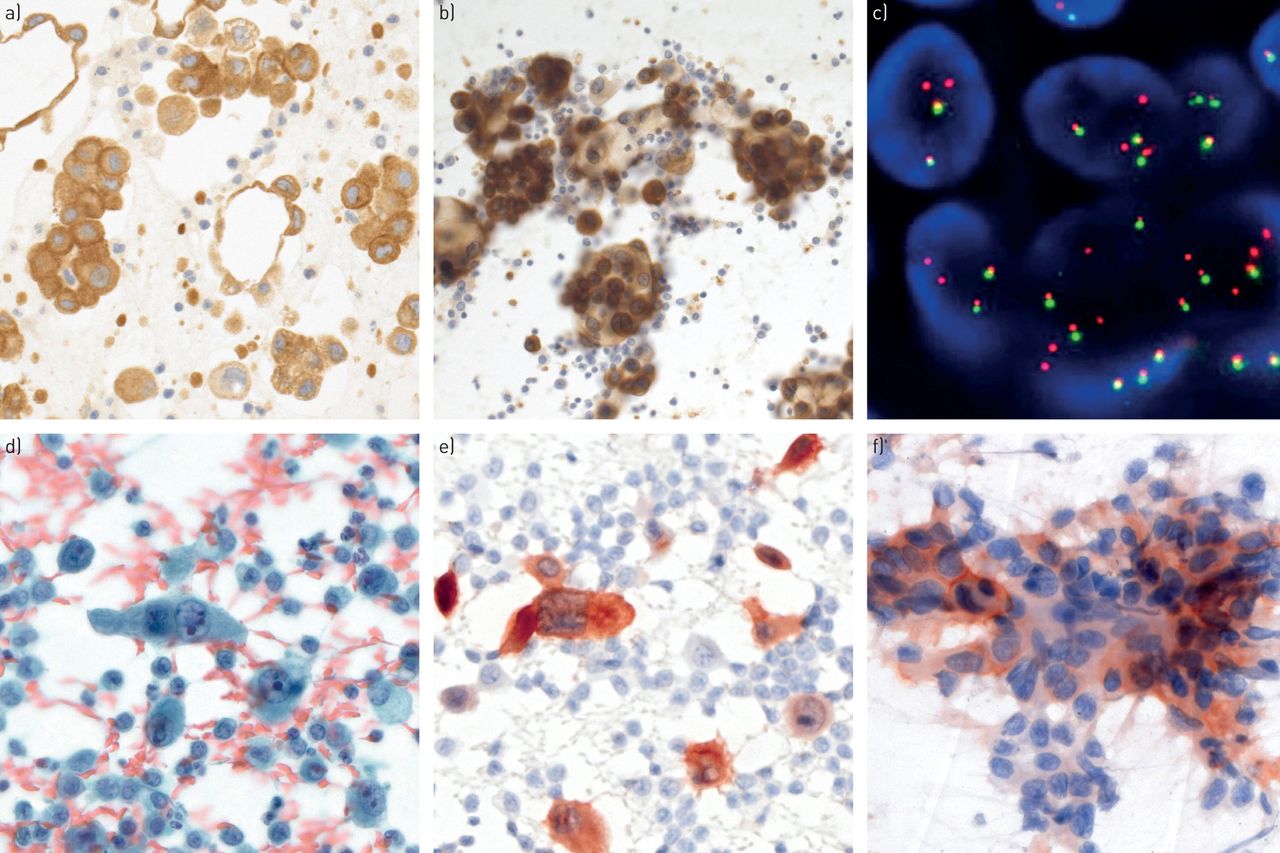
Predictive biomarker analyses in cytological specimens. a) Anaplastic lymphoma kinase (ALK) immunohistochemistry (IHC) on a formalin-fixed paraffin-embedded cell block and b) corresponding ethanol-fixed and previously Papanicolaou-stained cytological smear of a pulmonary adenocarcinoma (pleural effusion; Leica 5A4 antibody (Leica Biosystems, Newcastle upon Tyne, UK) on Ventana Benchmark XT (Tucson, AZ, USA) and Leica Bondmax automated immunostainers, respectively). c) ALK rearrangement shown by fluorescence in situ hybridisation (FISH) (two single red signals without corresponding green signals and two normal fusion signals per cell nucleus; break-apart FISH probe (Abbott Molecular, Abbott Park, IL, USA)). d) Papanicolaou-stained smear of ROS1-positive adenocarcinoma and e) corresponding ROS1 IHC (pleural effusion; Cell Signaling D4D6 antibody (Danvers, MA, USA) on Leica Bondmax automated immunostainer). f) Ethanol-fixed and previously Papanicolaou-stained smear with membranous and cytoplasmic positivity for programmed death ligand-1 positivity in a fraction of tumour cells (endoscopic ultrasound guided transbronchial fine-needle aspiration; Ventana SP142 antibody on Leica Bondmax automated immunostainer). a), b), d), e), f) Original magnification ×400; c) original magnification ×1000.
Thanks to the robustness of DNA, FISH and mutation testing is applicable to almost all types and formats of cytologic specimens irrespective of fixation type [114, 115]. As in IHC, cell blocks appear to be the most commonly used cytology format for FISH and mutation testing. However, not all cell blocks are adequate for predictive marker testing. Therefore, FISH analysis of smears/cytopsin/LBC (e.g. ALK or ROS1) should be considered as an option, and is preferred by some laboratories [110, 116, 117]. An important advantage of conventional cytology is the ability to select the most representative cytological slide among all previously stained slides. In addition to a lack of nuclear truncation and related artefacts, the DNA quality in alcohol-fixed cytological specimens is better than that after formalin fixation, allowing for brilliant technical FISH results. This explains the success rate of up to 100% for ALK FISH analysis in conventional cytology, compared to a failure rate of up to 19% for histological specimens and cell blocks [57, 110, 115]. Nevertheless, standardisation remains crucial, as highlighted by a recent study where 23% of 115 cytological specimens with heterogeneous pre-analytical conditions were not evaluable by ALK FISH [118]. Ideally, adhesive-coated or positively charged slides should be used to prevent the cells from floating off during the technical procedure. There may be concerns about sacrificing diagnostic cytological slides for FISH or mutation analysis because cytology laboratories are required by law to archive diagnostic slides in most European countries (for a variable number of years: UK 10 years, Germany 20 years and Belgium 30 years). These concerns can be addressed by capturing representative images or by scanning the whole slide before the additional analysis.
The utility of cytological specimens for mutation testing is now unquestioned with smears/cytospins/LBC specimens appearing as suitable as cell blocks [14, 119, 120]. This is true for all mutation testing methodologies, including panel testing using NGS, which is becoming a standard because of the necessity of testing for a growing number of targetable mutations [121, 122]. As in histological specimens, the enrichment of tumour cells for DNA extraction is required in specimens with an estimated fraction below a threshold defined by the method (e.g. 20% in case of NGS). This can be achieved by encircling areas with a high tumour cell proportion for manual macro- or microdissection. In prestained smears/cytospins/LBC specimens with a low tumour cell proportion, laser capture microdissection operated by trained cytotechnicians is another robust method for tumour cell enrichment that saves specimens that would otherwise not qualify for mutation testing [123, 124]. NGS lung panel testing using the Ion Torrent PGM (Life Technologies, Carlsbad, CA, USA) platform requires as few as 100–200 tumour cells collected from ethanol-fixed smears/cytospins/LBC [14].
The ion torrent personal genome machine (PGM) platform was selected for solid tumour testing primarily because of the low 10-ng input DNA requirement, which can be easily achieved from a modest 1000- to 1500-cell sample. In contrast, the input DNA requirement for the Illumina MiSeq (Illumina, San Diego, CA, USA) platform is 250 ng and requires between 5000 and 15 000 cells per sample for successful testing. The superior quality of DNA in ethanol-fixed cytology might explain why this material has been found to be more efficient for DNA extraction and mutation analysis than FFPE cell blocks [125].
Taken together, in this era of personalised medicine, cytology plays an important role in the management and prioritisation of tumour material for multiple predictive marker testing. However, further efforts are needed to standardise the pre-analytical procedures and protocols, and to ensure rigorous and continuous quality control.
Tissue management
The extended diagnostic and predictive requirement from increasingly limited material provided by minimally invasive biopsy and cytology techniques poses major challenges for pathology. Therefore, tissue management is essential: tissue sample size should be maximised whenever feasible [126]. In addition, tissue handling, processing and sectioning should be standardised to minimise wastage and optimise tissue usage for staining procedures and PCR-based molecular tests. Tissue management is an interdisciplinary challenge (figure 5) [127], as follows.
It starts with the physician (e.g. pulmonologist, radiologist, oncologist or surgeon) taking the sample in a patient with suspicion of pulmonary malignancy. The physician should fill the request form: “diagnosis? If malignant, predictive testing (D+P)”. This “D+P” directs the laboratory to perform careful, contaminant-free cutting of a FFPE block of ∼15 sections. The first and last section are stained with H&E. In cases of NSCLC-NOS some of the middle sections can be used for diagnostic stains (e.g. TTF1, P40 or mucin). In cases of adenocarcinoma or SqCC the predictive analysis can be performed on the remaining spare sections (ALK and ROS1 IHC or DNA/RNA extraction).
The superficial cutting initiated by the “D+P” code contrasts with the regular cutting procedure in daily pathology practice. With regular cutting, series of H&E sections from different levels in the biopsy (superficial, 100 µm deeper) are placed on one microscopic slide. This conventional approach provides confidence that it is representative of the disease state. However, in the great majority of positive biopsy samples the tumour tissue is visible in the superficial H&E sections. It is noteworthy that with superficial cutting no tissue is lost, so if the initial H&E sections do not contain tumour, deeper cutting is still possible. Generally, after the careful cutting of the FFPE block, sufficient tissue is left in the biopsy for additional DNA/RNA analysis if needed. It is important to know that replacing the FFPE block in the cutter for cutting new sections leads to loss of an essential part of the biopsy by the process of new aligning and straightening the first series of sections. In the context of research projects requiring extra tissue, additional sections for this purpose are best planned at the initial cutting process.
Once the diagnosis of NSCLC is made the “D+P” statement has informed the pathologist that predictive testing is mandatory and reflex testing should be initiated. Sections for predictive IHC (e.g. ALK and ROS1) and FISH (e.g. RET and MET) may be performed. Thus, the D+P code encompasses the agreement for reflex testing.
Whenever possible, the process of sampling should not be limited to one small sample. In the case of a visible tumour at bronchoscopy, preferably six biopsies should be taken and distributed over two or three vials with fixative. In the laboratory, this will translate into two or three FFPE blocks, which, depending on the tumour content, may be used separately for predictive analysis. During the process of transthoracic needle biopsy three needle biopsies should be taken and distributed over different vials. In case of sampling a cervical lymph node a surgical incisional or excisional biopsy is preferred over a needle biopsy, and FNA should be considered in addition. During rapid on-site evaluation by cytology a suspected site may be diagnosed with cancer for staging. However, the endoscope at the proper position should subsequently be used to perform more passes of the tumour-containing lesion and after every pass, the needle should be emptied into a preservative solution for preparation of a cell block. A cell block with a high tumour cell content and extra ethanol-fixed smears are as suitable as tissue samples. For pleural fluid with malignant cells, collecting a repeat puncture may increase the diagnostic sensitivity, but also provides more tumour cells to prepare extra smears and a representative cell block. Thus, sampling more of the tumour is always better than less, and the “D+P” code stimulates the preparation of extra specimens, including cell blocks.
A morphology that is unusual for a pulmonary adenocarcinoma may prompt the pathologist to consider a metastasis of a extrapulmonary primary tumour (e.g. breast, colon or kidney) and perform appropriate IHC stains in this direction, irrespective of clinical information (i.e. oestrogen, progesterone receptor and GATA3 for breast; CK20 and CDX2 for colorectal; or CD10 and PAX8 for kidney). However, unless the morphology raises such suspicion of metastasis, or if the sample request form does not contain any information of another known cancer or a clinical suspicion of metastatic disease, such extra IHC investigations should not be performed, as this can waste tumour material and jeopardise predictive marker testing.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Tissue management interaction between the sample collector and the pathologist. Reproduced and modified from [127] with permission.
Turnaround time
A morphologic diagnosis can be obtained in 1–3 days, depending whether or not diagnostic IHC is needed. When reflex testing is in place the predictive analysis can be initiated immediately after diagnosis. The turnaround time of the predictive analysis depends on the frequency of testing. The requirement for EGFR testing in lung cancer currently accepted is usually fewer than five working days, with a maximum of 10 working days. However, if EGFR/NGS analysis is performed once a week, this may add up to five working days. This may be reduced to up to three working days if NGS is performed twice a week. If a block needs to be shipped to another centre, a delay of several days is not exceptional. Once the block is in the laboratory the sample can be analysed by targeted NGS within five or six working days. Analysis for fusion genes performed sequentially in cases without an NGS-detectable predictive biomarker will add another few days for RNA-based gene fusions detection or FISH. Initiating IHC for ALK or ROS1 simultaneously with mutation testing allows the identification of the 6–7% of patients who are ALK- or ROS1-positive without delay. For clinical management, provisional results may be communicated in three steps: diagnosis; ALK and ROS1 IHC status if positive; and PD-L1 NGS and FISH for fusion analysis in case the other tests have been negative.
According to guidelines, the turnaround time for EGFR and ALK testing should be within 10 working days [16, 49]. In practice this is not always the case.
In conclusion, in this era of personalised medicine, small samples play an important role in the management and prioritisation of tumour material for multiple predictive marker testing in NSCLC. In general, it is preferable to obtain the biggest biopsy possible, taking into consideration patient risk and comfort.
Disclosures
Supplementary Material
L. Bubendorf ERR-0007-2017_Bubendorf
Footnotes
Previous articles in this series: No. 1: Ghigna MR, Mooi WJ, Grünberg K. Pulmonary hypertensive vasculopathy in parenchymal lung diseases and/or hypoxia. Eur Respir Rev 2017; 26: 170003.
Conflict of interest: Disclosures can be found alongside this article at err.ersjournals.com
Provenance: Commissioned article, peer reviewed.
- Received January 11, 2017.
- Accepted March 15, 2017.
- Copyright ©ERS 2017.
ERR articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










