





Abstract
Between September 2015 and August 2016 there were >1500 publications in the field of diffuse parenchymal lung diseases (DPLDs). For the Clinical Year in Review session at the European Respiratory Society Congress that was held in London, UK, in September 2016, we selected only five articles. This selection, made from the enormous number of published papers, does not include all the relevant studies that will significantly impact our knowledge in the field of DPLDs in the near future. This review article provides our personal view on the following topics: early diagnosis of idiopathic pulmonary fibrosis, current knowledge on the multidisciplinary team diagnosis of DPLDs and the diagnostic role of transbronchial cryobiopsy in this diagnostic setting, insights on the new entity of interstitial pneumonia with autoimmune features, and new therapeutic approaches for scleroderma-related interstitial lung disease.
Abstract
Current research is redefining disease boundaries, focusing on less invasive and early diagnosis of DPLDs http://ow.ly/zMdj30aADFz
Interstitial lung abnormalities: why we should care
Recent studies performed by computed tomography (CT) evaluation in family members of patients with familial idiopathic pulmonary fibrosis (IPF) [1], in healthy elderly patients [2] and, more recently, in large longitudinal cohorts for cardiovascular disease and lung cancer screening, provide proof of evidence for the early identification of subclinical interstitial lung abnormalities (ILAs) and set the conceptual basis for a possible screening strategy in diffuse parenchymal lung diseases (DPLDs). These advances have become possible by integrating the wide-scale use of high-resolution computed tomography (HRCT) in large longitudinal studies and due to the more recent advances in molecular medicine, particularly in relation to genetics. The MUC5B genotype, well known to be associated with a mild form of IPF, has been shown recently to be associated with the serial progression of ILAs, thus providing strong indirect support for the view that screening for ILAs might eventually provide a means for the early identification of IPF. Further molecular studies will elucidate the molecular profile of IPF, and eventually of ILAs, and this may lead in the future to the identification of different subtypes of pulmonary fibrosis for which the identification and treatment of early subclinical forms might become a reality.
Putman et al. [3] have elegantly shown that ILAs are associated with a greater risk of all-cause mortality and a higher rate of death from respiratory failure and pulmonary fibrosis. These findings support the view that ILAs may, in some cases, represent a pre-clinical form of pulmonary fibrosis, thus opening a new intriguing scenario in which a very early diagnosis of IPF and earlier treatment could potentially improve the long-term clinical outcome of this progressive and ultimately fatal disease.
ILAs are defined as nondependent changes affecting >5% of any lung zone, including reticular or ground-glass abnormalities, diffuse centrilobular nodularity, non-emphysematous cysts, honeycombing or traction bronchiectasis (figure 1) [3]. Previous studies have reported a prevalence of ILAs of ∼2–10% among patients enrolled in clinical trials [4–7] and 7% of the general population [7]. ILAs share with IPF and familial interstitial pneumonia a similar lung function profile (restrictive defects, reduced exercise capacity, gas exchange) [4, 5, 8] and genetic abnormalities [7]. Given these correlations between IPF and ILAs, Putman et al. [3] hypothesised that ILAs could represent the pre-clinical form of IPF and that the presence of ILAs would be associated with an increased rate of mortality. Their study was designed to address all-cause mortality of ILAs over a period of ∼3–9 years.
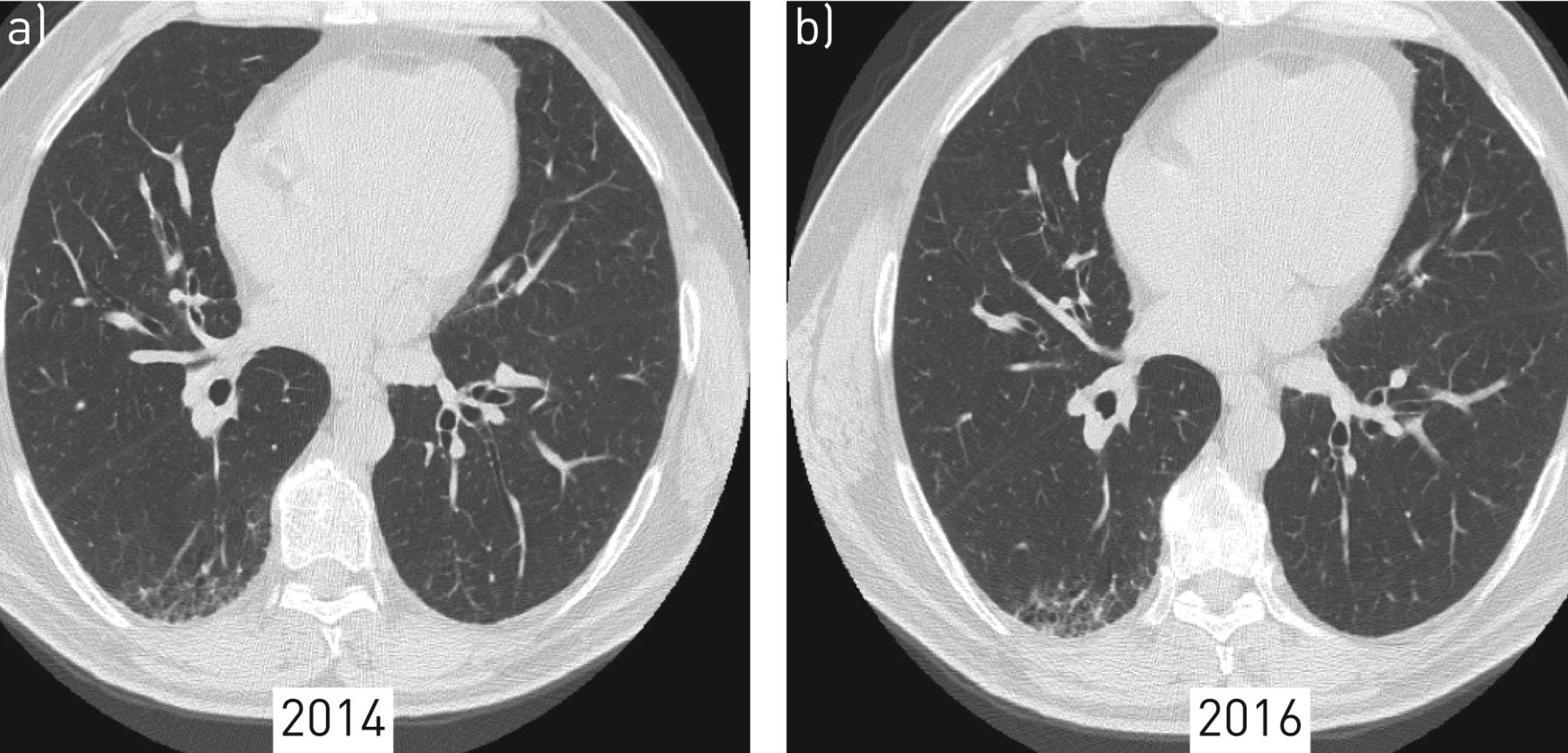
High-resolution computed tomography (HRCT) features of interstitial lung abnormalities. a) HRCT of a 56-year-old patient whose mother died of idiopathic pulmonary fibrosis at the age of 59 years. The patient sought our attention in 2014 when he was asymptomatic with normal pulmonary function test (forced vital capacity 140% of predicted, diffusing capacity of the lung for carbon monoxide 95% of predicted). The HRCT showed reticular changes involving <10% of the lung. b) After 2 years of follow-up, the HRCT shows slow progression, with more prominent architectural distortion and a few cysts compatible with early honeycombing.
Putman et al. [3] selected a large number of participants (11 691) from four studies: 2633 from the Framingham Heart Study (FHS; 2008–2011), a study designed to identify risk factors for cardiovascular diseases in the general population [9]; 5320 from the Age, Gene/Environment Susceptibility (AGES)–Reykjavik study (2002–2006), a longitudinal birth cohort from the observational Reykjavik Study [10]; 2068 from the COPDGene study (2007–2010), a study designed to identify risk factors for chronic obstructive pulmonary disease (COPD) (all smokers, with and without COPD) [5]; and 1670 from the Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) study (2005–2006; 500 were control smokers and nonsmokers) [11]. The prevalence of participants with ILAs in the four studies was 7% (n=177) in the FHS, 7% (n=378) in the AGES–Reykjavik study, 8% (n=156) in the COPDGene study and 9% (n=157) in the ECLIPSE study. Across all cohorts, ILAs were associated with older age when compared to participants without ILAs. For all cohorts, except that from the COPDGene study, the absolute mortality rates were significantly higher among participants with ILAs, resulting in a 6% difference (95% CI 2–10%) in the FHS, a 23% difference (95% CI 18–28%) in the AGES–Reykjavik study, a 5% difference (95% CI −1–11%) in the COPDGene study and a 6% difference (95% CI 1–11%) in the ECLIPSE study. In multivariable Cox proportional hazards models adjusted for age, sex, race, body mass index (BMI), pack-years of smoking, current smoking status and Global Initiative for Chronic Obstructive Lung Disease (GOLD) stage (where available), ILAs were associated with a higher risk of death in the FHS (hazard ratio (HR) 2.7, 95% CI 1.1–6.5; p=0.03), AGES–Reykjavik study (HR 1.3, 95% CI 1.2–1.4; p<0.001), COPDGene study (HR 1.8, 95% CI 1.1–2.8; p=0.01) and ECLIPSE study (HR 1.4, 95% CI 1.1–2.0; p=0.02). The association between ILAs and mortality remained statistically significant after additional adjustments for the percentage of emphysematous lung, reports of malignancy and measures of coronary artery disease (CAD), except in the FHS and COPDGene study, in which the adjustment for CAD resulted in no association. To determine the causes of death, Putman et al. [3] analysed the death certificates on an interim follow-up date (December 31, 2009; median follow-up time 5.4 years) from the AGES–Reykjavik cohort. Patients with ILAs were more likely to die of a respiratory cause (13%) than those who did not have ILAs (4%) or those with indeterminate status (6%; indeterminate status was defined by the presence of focal or unilateral ground-glass attenuation, reticulation or ground-glass abnormalities involving <5% of any lung zone on chest CT scan). After adjusting for covariates (age, sex, race, BMI, pack-years of smoking, current smoking status), participants with ILAs had a higher odds ratio (OR) of death from a respiratory cause (OR 2.4, 95% CI 1.7–3.4; p<0.001). ILAs were also associated with an increased rate of death from pulmonary fibrosis (seven (47%) out of the 15 respiratory deaths).
These findings, and those published previously [2–9], demonstrate that, despite often being undiagnosed and asymptomatic, ILAs are associated with lower survival rates and a higher risk of death from respiratory causes [1]. In three previous studies the prevalence of ILAs ranged from 3% to 10%, with estimates of progression ranging from 20% at 2 years to 46% at 4 years [6, 12, 13]. In a recently published study by Araki et al. [14], the results were comparable: ILAs were prevalent in 6% of total cases, and in 3% (n=53) of the initial scans, of which 43% (n=23) demonstrated progressive imaging abnormalities over ∼6 years of follow-up time. Consistent with previous reports, the progression of ILAs to imaging findings consistent with usual interstitial pneumonia (UIP) was seen in 4% of cases. ILAs have been defined in the majority of those studies as bi-basal reticular changes (around 80% of ILA cases showed this pattern in the study by Araki et al. [14]), with a large minority that progressed and, for those who progress, a significant increase in the risk of death (HR 3.9, 95% CI 1.3–10.9; p=0.01). Among 970 individuals without ILAs on first HRCT evaluation, 95 developed new ILAs after ∼6 years of follow-up, with 13% (n=12) showing definite fibrosis and 1% (n=1) showing UIP on follow-up HRCT. Based on these preliminary findings, we can conclude that ILAs are associated with an increased risk of pulmonary function decline and death, but the clinical profile remains elusive. Rather than being one entity, ILAs currently comprise an amalgamation of different subclinical DPLDs from different studies [15]. With the exception of the small proportion of UIP cases (1–4%), the remaining ILAs may encompass various subtypes of DPLDs, including smoking-related abnormalities. Histological and molecular profiling of ILAs is urgently needed in order to understand which cases of ILA represent the pre-clinical form of IPF. Moreover, the mortality rates associated with ILAs are lower than the mortality rates for IPF [16], and there is a large discrepancy between the prevalence of ILAs reported in this study (7% in the general population) and the known prevalence of IPF (0.04% of the general population) [16]. This may be due to the diagnostic delay for IPF and to there being a large prevalence of undiagnosed cases, or it may simply reflect the fact that ILAs represent the pre-clinical phase of IPF only in a small subgroup of patients that we are currently unable to identify.
Future studies are needed to elucidate when ILAs are clinically relevant. The presence of a family history for interstitial lung disease (ILD) and younger age may be relevant to discriminating pre-clinical IPF from the normal variant of the ageing lung observed in the elderly. Transbronchial cryobiopsy (TBCx), a novel mini-invasive tool for the histological diagnosis of DPLDs, offers the opportunity to biopsy a wide range of patients [17], and its application in the early diagnosis of DPLDs is of undoubted interest (figure 2). Molecular medicine can provide useful insights, helping to stratify patients with early and asymptomatic disease, and discriminating between different subgroups with different prognoses and treatment responses (e.g. MUC5B, “telomeropathies”, surfactant protein mutations). At present, the treatment and follow-up in early cases of DPLDs are not standardised, and future studies and guidelines should focus on this subgroup of patients, which is currently largely under-recognised. The study by Putman et al. [3] strongly supports the view that future clinical trials should be designed to include and characterise, clinically and morphologically, the early and asymptomatic stages of DPLDs.
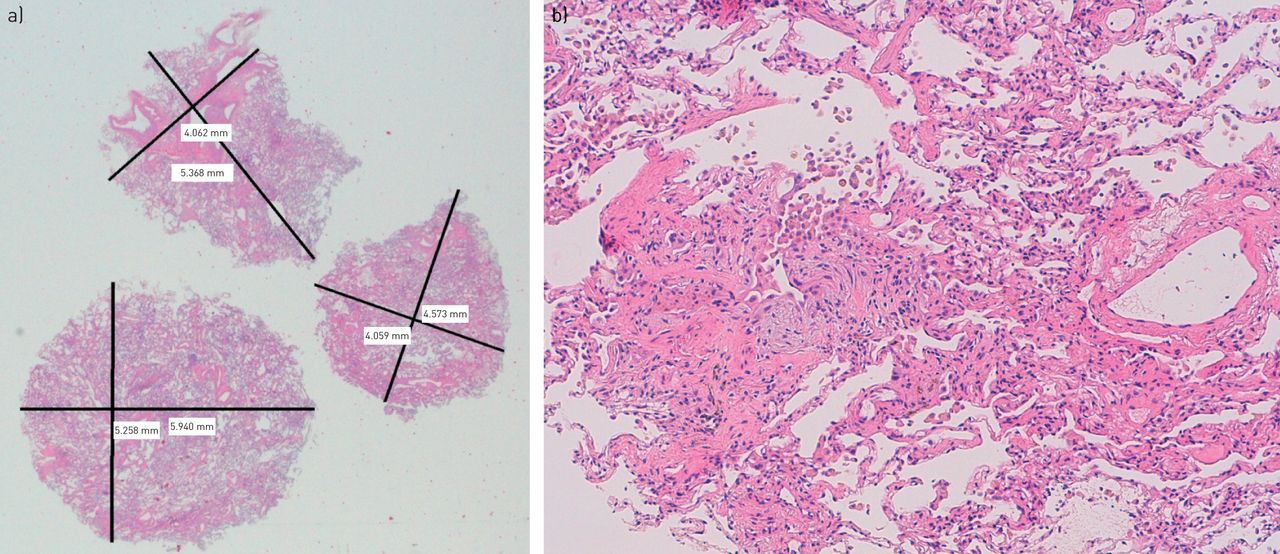
Transbronchial cryobiopsy (TBCx). a) Three fragments of TBCx measuring 18 mm2, 21 mm2 and 31 mm2 (total area of 70 mm2; low power of magnification, 4×). Biopsy was taken from the lower lobe of the right lung. The clinical case is briefly reported in figure 1. b) TBCx (high power of magnification, 40×) shows architectural distortion, patchy fibrosis and fibroblastic foci. Based on current American Thoracic Society/European Respiratory Society guidelines [16], pathologists identified a pattern of usual interstitial pneumonia with high confidence. This case of interstitial lung abnormalities is an example of a very early case of familial idiopathic pulmonary fibrosis. TBCx offers the opportunity to biopsy a wide range of patients [17], and its application in the field of early diagnosis of diffuse parenchymal lung diseases is of undoubted interest. However, treatment and follow-up of these patients are currently not standardised, and future studies and guidelines should focus on this subgroup of patients, which is currently largely under-recognised.
Multidisciplinary diagnosis of DPLDs: all that glitters is not gold
With the 2014 licencing of two new anti-fibrotic IPF drugs (pirfenidone [18] and nintedanib [19]), accurately diagnosing IPF, and discriminating it from the other fibrotic DPLDs, are of critical importance in clinical practice. Since the 2002 American Thoracic Society (ATS)/European Respiratory Society (ERS) joint statement [20], the multidisciplinary diagnostic approach (involving the integration of clinical, radiological and, in selected cases, pathological data) has been used for the diagnosis of DPLDs in several studies [21–24], and this has been re-stated as the diagnostic gold standard for DPLDs in the last revision of the ATS/ERS statement, published in 2013 [25]. Despite its recommended use, the multidisciplinary approach does have some relevant limitations: it is an imprecise process, it is difficult to standardise, it is inherently subjective, and there is no clear method to assess the accuracy of these multidisciplinary diagnoses. Moreover, significant disagreement exists in the multidisciplinary diagnosis of DPLD between physicians based in communities and those in academic centres. Flaherty and co-workers [23] showed higher levels of agreement between academic physicians, while among community clinicians, those who devoted the greatest time to the management of DPLDs showed higher agreement with their academic counterparts. This suggests that more intense interaction between academic and community physicians could improve diagnostic agreement between them. We acknowledge some limitations of this older study, which pre-dates the last revision of the ATS/ERS statement, and we recognise that fresh studies on this topic are highly desirable. However, the available body of evidence highlights the need to refer patients to dedicated centres, which may help to clarify diagnoses and provide suggestions regarding treatment options. Two simple but clinically highly relevant questions have remained unanswered in the last decade. 1) Are different expert centres “on the same page” when evaluating patients with suspected DPLDs? 2) Does the multidisciplinary diagnosis provide a correct prognostic stratification of patients? Walsh et al. [26] provide answers, evaluating, first, the level of agreement between international multidisciplinary teams (MDTs) of experts and, second, whether each team diagnosis correlates with mortality.
As reported by Walsh et al. [26], seven MDTs consisting of at least one clinician, radiologist and pathologist, from seven countries (Denmark, France, Italy, Japan, the Netherlands, Portugal and the UK), evaluated 70 cases of DPLDs in a two-stage process: 1) each observer independently evaluated each case and expressed their diagnostic likelihoods; 2) after multidisciplinary revision of all data, the MDT again chose their diagnosis likelihoods. In total, 18% (88 of all 490 MDT first-choice diagnoses) were IPF. Inter-MDT agreement for first-choice diagnoses was moderate overall (Cohen's kappa coefficient (κ) of 0.50). Inter-MDT agreement on diagnostic likelihoods was good for IPF (weighted kappa coefficient (κw) of 0.71, interquartile range (IQR) 0.64–0.77) and connective tissue disease (CTD)-related ILD (κw=0.73, IQR 0.68–0.78), moderate for nonspecific interstitial pneumonia (NSIP; κw=0.42, IQR 0.37–0.49) and fair for hypersensitivity pneumonitis (HP; κw=0.29, IQR 0.24–0.40). A total of 70% (347 out of 490) of first-choice MDT diagnoses were made with high confidence (>65% likelihood). High-confidence diagnoses of IPF were given in 68 (77%) out of 88 cases by MDTs, in 62 (65%) out of 96 cases by clinicians and in 57 (66%) out of 86 cases by radiologists. In six of the 15 cases in which the pathologists gave a first-choice diagnosis of IPF, the final MDT diagnosis was not IPF. Greater prognostic separation was shown for an MDT diagnosis of IPF than with individual clinicians’ diagnosis of this disease in five out of seven MDTs and radiologists’ diagnosis of IPF in four out of seven MDTs. The same analysis for pathologists' diagnosis of IPF was not significant for five out of seven pathologists, probably because of the small subgroup size (22 patients) and low prevalence of IPF in this subgroup.
Three previous studies evaluated the impact of “multidisciplinarity” in the diagnosis of DPLDs [24] and the agreement between different centres in the diagnosis of DPLDs [21, 23]. All three studies pre-dated the 2011 ATS/ERS consensus on IPF diagnosis and the 2013 revision of the DPLD ATS/ERS joint statement. The study by Walsh et al. [26], in line with previous results, confirms an acceptable level (κ>0.40) of diagnostic agreement between MDTs in the setting of DPLDs. Given that the distinction between IPF and other fibrotic DPLDs is of great clinical relevance for correct patient management, it is reassuring to note that inter-MDT agreement for the diagnosis of IPF was good (κ=0.60). Interestingly, clinicians had only marginally lower levels of agreement than MDTs for IPF diagnosis (κ=0.59), and, on subgroup analysis in patients without surgical lung biopsy, inter-MDT agreement and inter-observer agreement between clinicians for the diagnosis of IPF improved and were, again, almost the same (κ=0.70 and κ=0.71, respectively) [26]. These data suggest that current diagnostic criteria for IPF are clear and, for expert clinicians, are relatively easily applied even without the MDT discussion. MDTs are probably of greater importance in more complex cases in which clinical-radiological data are unclear or incongruent, and in this setting, lung biopsy adds an important piece of information. By contrast, non-IPF DPLDs are not well covered by evidence-based diagnostic guidelines, and this generates uncertainty and diagnostic disagreement between different centres. This is well reflected in this study by Walsh et al. [26], in which, for the diagnoses of NSIP and HP, the level of disagreement between MDTs is significantly higher compared to IPF.
Transbronchial cryobiopsy for the diagnosis of DPLDs: is it a step forward?
As recently mentioned by Colby et al. [27], “any new test is controversial, and cryobiopsy is no exception: it is considered by some to be a step forward” [28] “and by others to be a step backward” [29]. Several studies have clearly shown that TBCx provides larger specimens with fewer artefacts (figure 2) than regular forceps transbronchial biopsies (TBBx). In our previously published series [30], we reported a sensitivity for UIP detected by TBBx of only 30% for expert pathologists, and these data have recently been confirmed in a study that found TBBx useful to reach a confident and accurate multidisciplinary diagnosis in only 20–30% of patients with DPLDs, with the majority of cases requiring surgical lung biopsy (SLB) to secure a definite diagnosis [31]. By contrast, TBCx specimens are largely comparable to SLB, although the TBCx diagnostic yield falls short of that for SLB (80% for TBCx versus >90% for SLB), so TBCx has the potential to remove the need for surgery for a large proportion of patients. This is important, because the complication rate and mortality from cryobiopsy is significantly lower (0.1%) [32] than that for SLB (1.7% for elective procedures) [33]. TBCx is currently the most promising alternative to SLB, but it carries inherent risks, and patient safety is of great concern. Moreover, the technique has not yet been standardised and currently there is a wide variability among different centres in the equipment and personnel used, indications and contraindications. This causes a considerable variability in the size of the specimens (between 10 mm2 and 64 mm2) [27], diagnostic yield (from 50% to >90%) [32] and complication rates. The rate of pneumothorax is relatively high: 10% in a large literature review of almost 1000 patients and 20% (60 out of 297 patients) in our series [32]. Although not captured by the literature, mild bleeding is frequently observed with TBCx; in our meta-analysis, moderate bleedings were reported in seven of 12 studies, with a pooled estimate of 12% [32]. With the preventive use of a Fogarty catheter we observed 13 (4.3% of 297) cases of moderate bleeding (unpublished observation) and no case of severe bleeding.
Currently, there are no published studies on the diagnostic accuracy of TBCx. For practical reasons and concerns about patient safety, trials performing consecutive TBCx and SLB in the same patient have not yet been completed and, given the high diagnostic yield and low complication rate of TBCx compared to SLB, seem unlikely to be proposed to patients in the future. Our group recently published a study [34] comparing TBCx and SLB in a consecutive series of 117 patients (58 TBCx and 59 SLB) with fibrotic ILDs (all without definite UIP on HRCT). We applied the methodology of Flaherty et al. [24] to estimate the diagnostic impact of TBCx for the diagnosis of IPF in the multidisciplinary discussion process. The TBCx and SLB cases were reviewed by six experts in random order, with the addition of clinical-radiological, bronchoalveolar lavage, biopsy and follow-up data in a stepwise fashion. We recorded the diagnostic impressions and confidence levels of participants at each step with the aim of comparing whether the change in those variables before and after the addition of biopsy information differed in the TBCx and SLB groups. The multidisciplinary diagnoses were similarly distributed in the two groups, with an IPF prevalence of 50% (29 out of 58) and 39% (23 out of 59) for TBCx and SLB, respectively (p=0.23). The prevalence of an IPF diagnosis made with a high level of confidence doubled after adding histological information to both groups, from 29% to 63% with TBCx (p=0.0003) and from 30% to 65% with SLB (p=0.0016). The initial diagnostic impression changed after biopsy in a significant minority of cases, with 17% of cases in the TBCx group and 19% of cases in the SLB group being reclassified as IPF. The kappa coefficient of agreement in the diagnosis of IPF compared with other fibrotic ILDs increased similarly in the two groups (0.84 for TBCx and 0.74 for SLB). In conclusion, both approaches significantly affect multidisciplinary diagnostic impressions and the confidence levels of observers.
The current body of evidence clearly indicates that, in cases of suspected IPF, the specificity and positive predictive value (PPV) of small biopsies (TBBx) for UIP are very high (92–100% specificity and 86–100% PPV) [30], but with a very low sensitivity (30%). In this context, TBCx, which is known to dramatically increase the sensitivity of TBBx (from 30% diagnostic yield for TBBx to 80% for TBCx), is of undoubted utility. We can all agree that TBCx is clearly useful to confirm the presence of UIP in cases of suspected/possible IPF. In suspected cases of non-IPF, particularly HP and NSIP, the diagnosis is much more challenging and the issue is more complex. In this setting, TBBx has no role, with a negative predictive value for a UIP diagnosis ranging between 46% and 55% [30].
Our data show that TBCx has a role, because it was found informative in the majority of cases discussed by the MDT. However, concerns revolve around the possibility of losing “relevant ancillary features” (e.g. granulomas or centrilobular fibrosis suggestive of HP) or “coexistent patterns” (e.g. mixed histological features of UIP and NSIP in the same patient) when using samples that are smaller than those from SLB. The most relevant step forward in the diagnosis of DPLDs would be to clarify the true clinical relevance and real prognostic impact of those “ancillary features” detected by SLB. Molecular medicine is clearly showing that many of the current boundaries separating different morphological entities are probably not so important. When it comes to pathogenic mechanisms, different pathological-radiological entities with the same molecular defect seem to behave similarly (e.g. telomeropathies have the same dismal prognosis, regardless of whether they look like NSIP, UIP or HP) [35]. The ultimate proof of the value of cryobiopsy in the setting of DPLDs will not be to show that it can detect the same amount of “ancillary features” currently detected by SLB, but will be to show that TBCx can stratify patients prognostically and can be reliably used for molecular analysis.
Clarifying the features of IPAF
In a recent ERS/ATS research statement [36], the term “interstitial pneumonia with autoimmune features” (IPAF) was proposed to describe cases of patients presenting with an “idiopathic” interstitial pneumonia (IIP) associated with features suggestive of, but not diagnostic for, a classical CTD. IPAF has been defined as a combination of features from any two of three domains: a clinical domain, consisting of specific extra-thoracic features; a serological domain, consisting of specific autoantibodies; and a morphological domain, consisting of specific chest imaging, histopathological or pulmonary physiological features. IPAF was created as a research category, intended to provide a sound platform for the prospective study of these patients. With the aim of characterising the features of IPAF and defining its prognosis in comparison to other DPLDs, Oldham et al. [37] applied IPAF criteria to a series of 422 patients with IIPs (NSIP, IPF, cryptogenic organising pneumonia (COP) and unclassifiable IIPs) and undifferentiated CTD (UCTD)-associated ILD. A large subgroup of patients (34%, n=144) met the IPAF criteria (30% of NSIP, 60% of COP, 20% of IPF, 25% of unclassifiable IIPs and 91% of UCTD-ILD). The most common clinical findings were Raynaud's phenomenon (27.8%), inflammatory arthritis/morning stiffness lasting >60 min (17.4%) and mechanic's hands (10.4%). Antinuclear antibody ≥1:320 (or nucleolar or centromere pattern of any titre) was the most common serological finding (77.6%), followed by anti-Sjögren's-syndrome-related antigen A (16.6%) and rheumatoid factor ≥2× the upper limit of normal (13%). An NSIP pattern by HRCT was found in 31.9% of patients, while histopathological NSIP and organising pneumonia (OP) patterns were found in 22.9% and 16.9% of patients biopsied, respectively. Intrinsic airways disease was the most common multicompartment finding (22.2%), followed by pleural disease (12.5%) and pulmonary vasculopathy (18.8%). Over 75% of NSIP-COP and IPF patients met IPAF criteria through the serological-morphological domain, with 22% of NSIP-COP patients also fulfilling the clinical-morphological domain (compared to 0% of IPF fulfilling the clinical-morphological domain). UCTD-ILD patients met IPAF criteria in all three domains in nearly 50% of cases. In univariate Cox regression, predictors of increased mortality risk among those meeting IPAF criteria included the following: age (HR 1.06, 95% CI 1.03–1.08; p<0.001), prevalent hypothyroidism (HR 1.97, 95% CI 1.05–3.67; p=0.03), UIP pattern on HRCT and/or SLB (HR 2.4, 95% CI 1.21–4.76; p=0.01) and diffusing capacity of the lung for carbon monoxide (HR 0.97, 95% CI 0.96–0.99; p<0.001). Multicompartment features significantly increased mortality, whereas the presence of the clinical domain was associated with a significantly decreased mortality risk (HR 0.56, 95% CI 0.32–0.96; p=0.03). IPAF cohort survival was worse than CTD-ILD (p<0.001) and marginally better than IPF (p=0.07). After stratifying the IPAF cohort based on the presence of a UIP pattern by HRCT or SLB, those with a non-UIP pattern demonstrated survival similar to those with CTD-ILD (p=0.45) and those with a UIP pattern demonstrated survival similar to those with IPF (p=0.51) (figure 3).

{kind=link}
{kind=link}
{kind=link}
Kaplan–Meier survival curves of interstitial pneumonia with autoimmune features (IPAF) with usual interstitial pneumonia (UIP) pattern (on high-resolution computed tomography and/or surgical lung biopsy), IPAF without UIP pattern, idiopathic pulmonary fibrosis (IPF) and connective tissue disease (CTD)-interstitial lung disease (ILD) cohorts. IPAF patients without UIP demonstrated survival similar to those with CTD-ILD (p=0.45), while those with UIP demonstrated survival similar to those with IPF (p=0.51). Reproduced from [37] with permission.
In conclusion, this study by Oldham et al. [37] is an important first step in evaluating the new IPAF classification criteria, providing useful insights into the characterisations of clinical, morphological and serological features of IPAF in NSIP-OP, IPF, unclassifiable IIPs and UCTD-ILD patients. The strongest message of this study is that the outcome difference in IPAF is primarily linked to the underlying presence of UIP [38], which shows a prognosis similar to IPF, and we believe should be managed accordingly until existing uncertainties are clarified by future studies.
New therapeutic strategies for scleroderma-related lung fibrosis
Lung fibrosis has emerged as a major cause of death in scleroderma, one of the most relentlessly progressive immune-mediated rheumatic diseases involving different internal organs. Trials of therapies for lung fibrosis in systemic sclerosis face several challenges, first because of its rarity, and second because of its heterogeneous outcomes, which make evaluation of any treatment compared with placebo very difficult. Building on the preceding Sclerodema Lung Study (SLS) I trial [39], Tashkin et al. [40] compared, in a double-blind multicentre phase III trial, 12 months of oral cyclophosphamide (CYC; daily dose of 1.8–2.3 mg·kg−1) with 24 months of oral mycophenolate mofetil (MMF; daily dose of 3 g). Based on the previous SLS I trial, the authors hypothesised that MMF would have a significantly greater effect on % predicted forced vital capacity (FVC) than CYC, with an expected effect size at 24 months of 4% improvement after adjustment for baseline FVC and HRCT-measured fibrosis score. The primary end-point of this courageous trial was not met. Each treatment group showed a significant increase in % predicted FVC from baseline at each time-point (12 months, 18 months, 24 months), which was greatest at 24 months, and the course of % predicted FVC during the entire 24 months did not differ between the two treatment arms (p=0.24) and no difference was observed in % predicted FVC change from baseline at 24 months (−0.70, 95% CI −3.1–1.7; p=0.56). The predominant adverse events were anaemia and leukopenia, which occurred in significantly more patients in the CYC group than in the MMF group (30 versus four patients; p<0.05). Thrombocytopenia, although infrequent, occurred exclusively in the CYC group (four versus zero patients; p<0.05). Numerically, more serious adverse events related to the study drug occurred in the CYC group (n=8) than in the MMF group (n=3).
Contrary to the primary hypothesis, no significant difference was noted between the two treatments in the course of % predicted FVC over 24 months, but haematopoietic suppression occurred more frequently with CYC than with MMF, thus leading to the conclusion that MMF is a safe and equally efficacious alternative to oral CYC for the treatment of scleroderma-related ILD (SSc-ILD). This study by Tashkin et al. [40] is in line with earlier retrospective datasets, and supports the currently quite common use of MMF in clinical practice for SSc-ILD. However, these results should be regarded with cautious enthusiasm. Substantial limitations of the study include the premature withdrawal of a significant proportion of participants over the 24-month treatment phase and the choice of oral CYC over intravenous CYC, which is known to be safer and better tolerated. Clearly, better therapies are still needed for the treatment of SSc-ILD. MMF might emerge as a background immunotherapy to which other drugs, targeting vasculopathy, the immunoinflammatory compartment and epithelial-mesenchymal dysregulation, might eventually be added [41]. This direction is indicated by the recently published results of the LOTUSS trial [42], an open-label, phase II study of the safety and tolerability of pirfenidone in patients with SSc-ILD. The study demonstrated that pirfenidone has an acceptable tolerability profile in SSc-ILD, showing that the tolerability profile is not affected by the concomitant use of MMF. This study provides a strong rationale for further investigation of pirfenidone in future clinical trials in patients with SSc-ILD and opens a favourable scenario for the use of combination therapy in SSc-ILD in the near future. There is also pre-clinical evidence for the efficacy of nintedanib in SSc-ILDs [43], and an ongoing trial (ClinicalTrials.gov identifier NCT02597933) is exploring the efficacy and safety of nintedanib 150 mg (twice daily) in treating patients with SSc-ILD, compared with placebo. Moreover, small but relevant studies have shown that rituximab may improve lung function in patients with SSc [44, 45], and B-cell depletion has been shown to downregulate type I collagen gene expression and intracellular signalling triggered by anti-platelet-derived growth factor receptor autoantibodies, reducing scleroderma fibroblast activation [46].
Conclusions
These five studies on DPLDs published between September 2015 and August 2016 have been chosen to underline some relevant evolving concepts in the field of DPLDs. First, regarding the importance of an early diagnosis of DPLDs, the study by Putman et al. [3] clearly shows that a subgroup of ILAs evolve in pulmonary fibrosis and lead to an increased mortality for respiratory failure, so our future efforts should be focused on redefining the clinical and molecular features of those cases, identifying those patients who can benefit from timely treatment. Second, the multidisciplinary diagnosis of DPLDs is confirmed by Walsh et al. [26] to be feasible (with good interobserver agreement for the distinction of IPF based on current international guideline criteria) and informative (providing prognostic information). Its role is probably more relevant in the interpretation of complex cases than for cases in which the clinical-radiological picture is congruent and straightforward. Most notably, there is a gap between our diagnostic ability in interpreting IPF cases (good agreement) and non-IPF cases (low agreement for NSIP and HP), thus highlighting the need for clarification and for updated consensus guidelines for non-IPF DPLDs. Third, when histology is required, TBCx can be helpful, providing meaningful information in the context of dynamic interactions between clinicians, radiologist and pathologists in a fashion similar to that of surgical lung biopsy [34]. TBCx is a safe and useful mini-invasive method for the diagnosis of UIP/IPF, but the ultimate proof of its value in the setting of DPLDs requires showing that it can stratify patients prognostically. Fourth, the clinical relevance of a diagnosis of IPAF when not meeting definite criteria for CTD remains controversial. The recent proposed classification of IPAF identifies, in patients without UIP, a subgroup of patients with a prognosis similar to CTD-ILD. However, patients with IPAF and UIP behave like those with IPF/UIP, and we propose to continue to manage those patients according to the current diagnostic criteria for IPF [16] until existing uncertainties about IPAF/UIP are clarified by future studies. Fifth, recent trials show that MMF is a safe alternative to oral CYC for the treatment of SSc-ILD, and there are no safety issues in the combination therapy of MMF and pirfenidone. These data, in line with previous reports, support the current use of MMF in SSc-ILD, and encourage clinical trials to investigate the efficacy of new combination therapy for the treatment of SSc-ILD.
Disclosures
Supplementary Material
V. Poletti ERR-0004-2017_Poletti
C. Ravaglia ERR-0004-2017_Ravaglia
S. Tomassetti ERR-0004-2017_Tomassetti
Footnotes
Conflict of interest: Disclosures can be found alongside this article at err.ersjournals.com
Provenance: Commissioned article, peer reviewed.
- Received January 6, 2017.
- Accepted March 1, 2017.
- Copyright ©ERS 2017.
ERR articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References











Jump To
- Article
- Abstract
- Abstract
- Interstitial lung abnormalities: why we should care
- Multidisciplinary diagnosis of DPLDs: all that glitters is not gold
- Transbronchial cryobiopsy for the diagnosis of DPLDs: is it a step forward?
- Clarifying the features of IPAF
- New therapeutic strategies for scleroderma-related lung fibrosis
- Conclusions
- Disclosures
- Footnotes
- References
- Figures & Data
- Info & Metrics