





Abstract
At least 10% of patients with interstitial lung disease present monogenic lung fibrosis suspected on familial aggregation of pulmonary fibrosis, specific syndromes or early age of diagnosis. Approximately 25% of families have an identified mutation in genes mostly involved in telomere homeostasis, and more rarely in surfactant homeostasis.
Beyond pathophysiological knowledge, detection of these mutations has practical consequence for patients. For instance, mutations involved in telomere homeostasis are associated with haematological complications after lung transplantation and may require adapted immunosuppression. Moreover, relatives may benefit from a clinical and genetic evaluation that should be specifically managed.
The field of genetics of pulmonary fibrosis has made great progress in the last 10 years, raising specific problems that should be addressed by a specialised team.
Abstract
Monogenic pulmonary fibrosis raises specific problems that should be addressed by a specialised team http://ow.ly/YJUM30aoREG
Introduction
A genetic predisposition to interstitial lung disease (ILD) is suggested by a 10-fold increase in the prevalence of the disease in families of patients with a diagnosis of idiopathic pulmonary fibrosis (IPF) [1, 2]. Genetic studies of familial forms of ILD led to the discovery of mutations in genes implicated in telomere homeostasis (TERT, TERC, RTEL1, PARN, DKC1, TINF2 and NAF1) or surfactant homeostasis (SFTPC, ABCA3 and NFKX2-1) or associated with complex syndromes (COPA, TMEM173, HPS-1 to HPS-8, NF1, FAM111B, NDUFAF6 and GATA2) (table 1 and figure 1) [3–11]. International guidelines on clinical practice for ILD are needed, but here we describe our current practice with the disease.
Main genes with rare variants associated with pulmonary fibrosis

a) Main genes and b) corresponding pathways identified in 2016 as a Mendelian cause of suspected monogenic pulmonary fibrosis in adults.
This review focuses on the genetics of pulmonary fibrosis in three sections: 1) a short review of the topic; 2) indications for genetic testing and current care of patients with ILD associated with an identified germline mutation; and 3) a discussion on the appropriate management for relatives.
Pulmonary fibrosis associated with genetic mutations
General characteristics of familial pulmonary fibrosis
Epidemiology and risk factors
The prevalence of IPF is estimated at one in 5000 people [12], whereas the prevalence of ILD in a first-degree relative of a patient with IPF is ∼10%, which indicates a familial linkage [1, 13–15]. The pedigree features an autosomal-dominant transmission in 80% of cases, with cases occurring in several successive generations (vertical transmission) without known consanguinity [2, 16, 17]. Not surprisingly, in a case–control Mexican study, family history of pulmonary fibrosis was a risk factor for IPF (OR 6.1, 95% CI 2.3–15.9) [15]. The risk of death is increased 4.7-fold for pulmonary fibrosis in first-degree relatives [18]. Also, the risk of developing hypersensitivity pneumonitis (HP) is increased in the same family: in a Japanese series of 114 patients with chronic HP, 17.5% had a family history of pulmonary fibrosis [19].
In ILD families, the presence of ILD is more frequent in men, smokers and older adults (mean age 68 years) [16].
Radiographic and pathological pattern and evolution
Two retrospective series reported a discordant prevalence of chest computed tomography (CT) pattern in familial pulmonary fibrosis (FPF) [16, 20]. Although one series of 289 FPF patients reported a prevalent unclassifiable radiological pattern (50%), followed by usual interstitial pneumonia (UIP) (22%), nonspecific interstitial pneumonia (NSIP) (12%) and organising pneumonia (1%), another series of 309 patients showed a prevalent pattern of UIP (80%), followed by unclassifiable (12%) and NSIP (6%).
Different pathological patterns may be observed in FPF, although UIP is the most frequent (25–50% of cases) [20, 21]. A pattern of NSIP, fibroelastosis, organising pneumonia or suggested HP may be observed. However, in a series of 30 cases re-evaluated by three pathologists, the histology pattern was unclassifiable in 17 [20, 21].
In most series, the evolution of FPF was comparable to that of IPF sporadic forms and the causes of death were similar to those observed with IPF sporadic forms [22–24]. As observed in sporadic pulmonary fibrosis, a definite radiological or histological UIP pattern is associated with poor prognosis in FPF [16, 21].
Asymptomatic involvement
Systematic high-resolution chest CT performed in asymptomatic members of families with FPF revealed ILD in 14–25% [2, 16, 25]. The most commonly observed anomalies were thickening of septal lines, peribronchovascular thickening, subpleural reticular opacities and ground-glass opacities [2, 16, 25].
Transbronchial biopsies from 71 asymptomatic relatives of FPF patients presented histological abnormalities in 26 (36.6%), including interstitial fibrosis (n=12, 16.9%), peribronchiolar fibrosis (n=15, 21.1%), chronic inflammation (n=10, 14.1%), respiratory bronchiolitis (n=2, 2.8%) or granulomas (n=6, 8.5%) [2].
Genetic abnormalities associated with pulmonary fibrosis
Telomerase complex mutations
Genetic mutations and complex inheritance
The telomerase complex catalyses the addition of repeated DNA sequences in the telomere region, thereby protecting chromosomes against loss of material during mitoses [26]. The activity of the telomerase complex requires the activity of several proteins, including telomerase reverse transcriptase (TERT), dyskerin (encoded by the DKC1 gene) and telomere binding protein (TIN2, encoded by the TINF2 gene), interaction with the telomerase repeat binding factor (TERF1) and the telomerase RNA component (TERC, encoded by the TERC gene, also known as TR or hTR), a specialised RNA that contains a template for telomere repeat addition [26]. Recently, in addition to the telomerase complex, other proteins, such as the regulator of telomere elongation 1 (RTEL1), poly(A) specific ribonuclease (PARN) and nuclear assembly factor 1 (NAF1), have been implicated in telomere maintenance and pulmonary fibrosis [5, 27].
Heterozygous mutations have been detected in familial forms of pulmonary fibrosis involving TERT (∼15%), RTEL1 (5–10%), PARN (∼5%) and TERC (∼3%). Mutations in DKC1, NAF1 and TINF2 are much rarer (figure 1) [9, 23, 28–35]. TERT or TERC mutations may be found in about 1–3% of sporadic IPF cases [28]. None of these genes is the site of a frequent mutation, and new genetic variants are continually being identified (e.g. as reported in the Telomerase Database, http://telomerase.asu.edu/diseases.html). The penetrance (risk of pulmonary fibrosis developing in a carrier of TERT and TERC mutations) depends on several factors, including environmental exposure [28, 36]. The gene–environment interaction reported for carriers of TERT or TERC mutations may also be implicated for carriers of RTEL1 mutations [9].
Patients with mutations in telomere-related genes show shortened telomeres in 80–90% of cases [28, 37]. Patients with ILD have shorter telomeres than controls, and patients with IPF have shorter telomere length than patients with other ILD. Within patients with FPF, patients with TERT mutations have the shorter telomere [37]. Telomere length may be measured on circulating leukocytes, and flow cytometry with fluorescent in situ hybridisation (flow-FISH), quantitative PCR and Southern blot are methods that are used for diagnostic purposes, but the most important factor for accurately defining telomere shortening is a well-defined control population [38]. Telomere length shortens with age, and the decrease is accelerated on exposure to toxins (tobacco, pesticides) and with numerous chronic diseases [39–42].
Telomeres shorten from generation to generation in patients with TERT, TERC or RTEL1 mutations because of transmission of the short telomeres, independent of transmission of the mutation [9, 28]. Phenocopy is defined in this review in a familial context of pulmonary fibrosis as an affected relative with a different genotype from the proband. This epigenetic-like inheritance in human disorders of telomerase explains the occurrence of phenocopies (an affected patient without the familial mutation) [17, 43] and the phenomenon of genetic anticipation, defined by an earlier onset of the disease with each generation (figure 2) [17, 43, 44], and this complicates genetic counselling. Because telomere shortening varies depending on the involved gene and its impact on telomere length, genetic anticipation may be more pronounced for carriers of TERC than PARN mutations [9].

Typical pedigree of a family suggestive of a TERC mutation with autosomal dominant transmission and anticipation. Ages are those reported at diagnosis. ILD: interstitial lung disease. The arrow indicates the proband.
Pulmonary involvement
The prevalence of ILD in carriers of gene mutations increases with age [28]. In an American cohort of TERT mutation carriers, no patients younger than 40 years showed ILD, but the prevalence was >60% in those older than 60 years [28]. Patients with TERC mutations may be relatively younger at ILD diagnosis than cases of mutations in other genes [9]. In a recent series of 114 patients with ILD associated with telomerase complex mutation, ILD was diagnosed at a mean age of 51 years (n=7) for TERC mutation carriers, 58 years (n=75) for TERT mutation carriers, 60 years (n=14) for RTEL1 mutation carriers and 65 years (n=19) for PARN mutation carriers (p=0.03) [9, 36]. The male/female ratio of patients with ILD varies from 0.5 to 0.7. In one series, men were younger than women at ILD diagnosis (age 54 versus 63 years) [28]. Smoking and pneumotoxic exposure are frequently observed in patients with ILD (40–96%) [9, 28, 36].
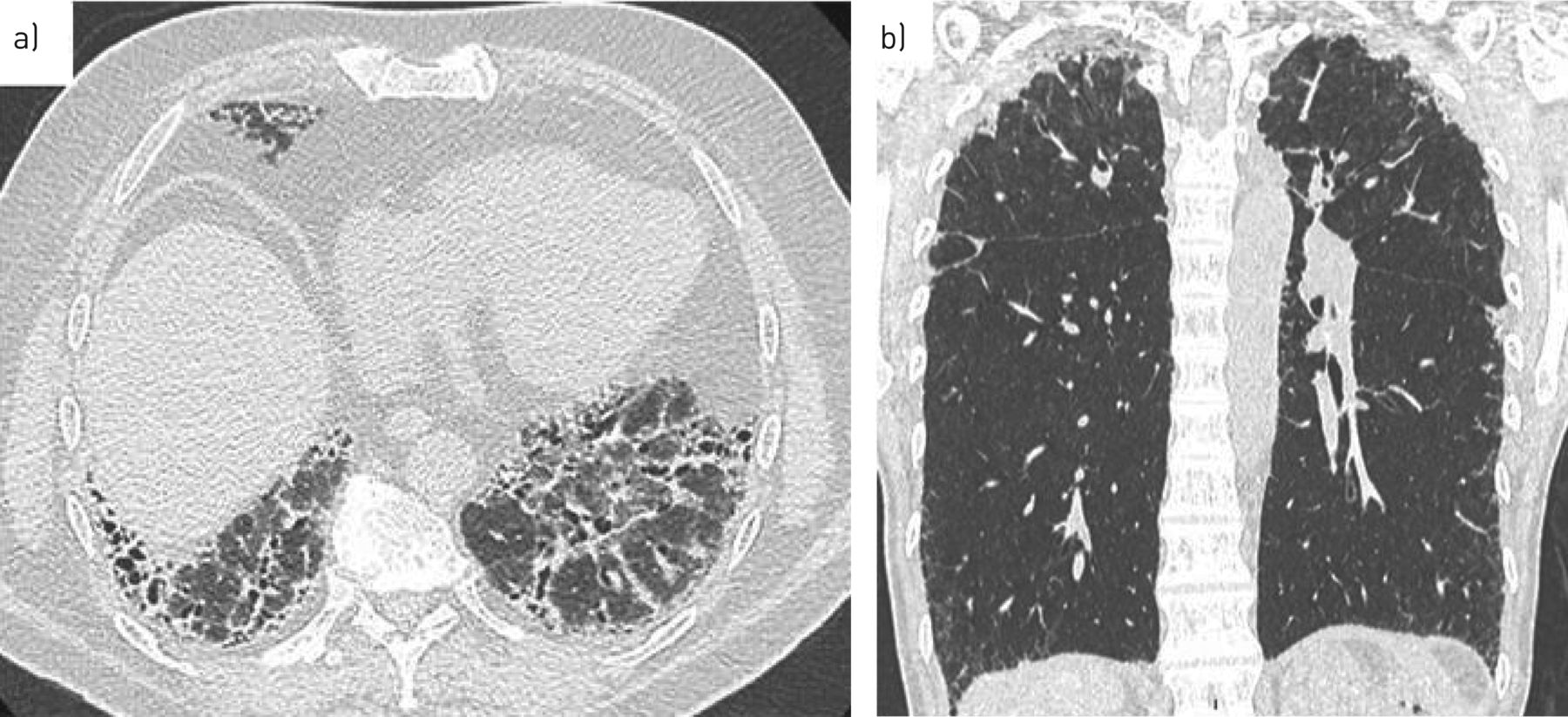
A typical UIP pattern on chest CT was initially reported in up to 74% of cases, but was recently found in only 46–55% of cases (figure 3) [9, 28, 36]. Unusual features found in 13–20% of cases included upper-lung predominance of fibrosis, centrolobular fibrosis, or a pleuro-parenchymal fibroelastosis pattern [9, 28, 36]. In all, 14–40% of cases show a combined pulmonary fibrosis and emphysema pattern [45]. TERT and NAF1 mutations could be specifically associated with severe chronic obstructive pulmonary disease and emphysema, particularly in female smokers, with increased risk of pneumothorax [5, 46].

Representative computed tomography scans of carriers of a) RTEL1 and b) TERC mutations. Usual interstitial pneumonia is the most frequent pattern seen (a), but a pattern suggestive of pleuro-parenchymal fibroelastosis diagnosis seems unexpectedly frequent (b).
IPF is the most frequent diagnosis (86% in one cohort, 45–47% in recent cohorts) [9, 28, 36]. Pleuroparenchymal fibroelastosis (up to 10%), chronic HP (7–11%) and unclassifiable fibrosis (19–30%) are also frequent [9, 36].
The decline of pulmonary function in patients with ILD associated with telomerase complex mutations seems unexpectedly high when compared to placebo arms of IPF clinical trials (decline of forced vital capacity (FVC) of 130–210 mL·year−1) [47]. Newton et al. [9] recently reported a 300 mL·year−1 decline of FVC, whatever the gene involved (TERC, TERT, RTEL1 or PARN) and the ILD entity (IPF or not).
Pulmonary fibrosis associated with TERT mutation is lethal, and most patients die of respiratory causes [9, 36]. The mean survival for symptomatic patients is 2.8–5.2 years after diagnosis. However, the evolution can be longer, particularly in asymptomatic patients [48]. Transplant-free survival could be lower for carriers of TERT or TERC mutations than patients with familial ILD without TERT or TERC mutation [36].
The safety and effectiveness of antifibrotic drugs has not been established in patients with genetic forms of pulmonary fibrosis. Our preliminary data in a cohort 18 patients with TERT or TERC mutations suggest that pirfenidone is well tolerated [49]. Very recently, danazol, a synthetic sex hormone with androgenic properties, showed promise for pulmonary fibrosis associated with telomere disease, with stabilisation of diffusing capacity of the lung for carbon monoxide (DLCO), FVC and CT scan findings during the 2-year treatment [50]. Moreover, treatment with danazol was associated with telomere elongation and haematological response in 79% of cases (19 out of 24). However, this drug is poorly tolerated in many patients, with liver adverse effects and increased risk of venous thrombosis. Drugs targeting telomere homeostasis, such as fluoxetine, cycloastragenol, resveratrol, N-acetylcarnosine or AGS-499, are being evaluated [51–57].
Given the young age of most patients, lung transplantation is often discussed. Three retrospective series recently reported the outcome of lung transplantation in 26 patients with TERT and TERC mutations [58–60]. Almost all patients required adjustment of immunosuppression because of haematological toxicity. Thrombocytopenia and a need for platelet transfusion were frequent, and myelodysplastic syndrome and/or bone-marrow failure occurred in some patients. Acute kidney failure requiring dialysis support seemed unexpectedly frequent in two series (0–50%) [58–60]. The prevalence of infections and acute/chronic rejections did not differ from historical data, but there was no control group. Some groups suggest performing bone-marrow biopsy for evaluation before lung transplantation and to adapt immunosuppression for selected patients [61].
Extrapulmonary manifestations
Pulmonary fibrosis and extrapulmonary manifestations associated with telomerase complex mutations are variably called “telomere disease”, “telomeropathy” or “short telomere syndrome”, with a lack of consensual definition.
Mucocutaneous involvement: Mutations in telomerase enzyme components were first identified in a rare syndrome called dyskeratosis congenita (DKC), the first telomeropathy described [62, 63]. International guidelines have recently been published [64]. DKC is classically defined by a triad of mucocutaneous manifestations: reticular skin pigmentation, nail dystrophy and oral mucosal leukoplakia [64]. The triad is present in childhood, and bone-marrow failure usually appears in the second decade of life [64]. Lung fibrosis is also a feature that may spontaneously appear in DKC, and these patients show severe pulmonary complications after haematopoietic stem-cell transplantation [65].
In patients with TERT mutation and pulmonary fibrosis, the classical triad of DKC is usually absent. However, 15–40% of mutation carriers present premature hair greying (age <30 years) [36, 66]. The same gene mutation can lead to different phenotypes: descendants from patients with pulmonary fibrosis and telomerase complex mutation may present typical DKC [9].
Haematological involvement: Telomerase complex mutations have been described in patients with bone-marrow failure, myelodysplasia or acute leukaemia in the absence of mucocutaneous DKC signs [67–69]. The coexistence of pulmonary fibrosis and bone-marrow failure in a given patient or a family with autosomal-dominant transmission highly suggests telomerase complex mutation [70]. However, myelodysplasia and IPF may both occur in patients aged >75 years without any telomerase complex mutation identified (personal observation).
In patients with telomerase complex mutations and pulmonary fibrosis, anaemia is present in 17–27%, macrocytosis in 24–41% and thrombocytopenia in 8–54% [9, 28, 36]. DKC1, TINF2 and TERC mutation carriers seem more prone to the development of haematological involvement than TERT or RTEL1 mutation carriers (personal observation and [9]).
Liver involvement: Patients can also present liver cirrhosis, which may be cryptogenic, viral or alcoholic. In a series of 10 patients with liver cirrhosis and TERT or TERC mutations, all were considered to have cirrhosis secondary to hepatitis C, alcohol consumption, Wilson's disease, nonalcoholic steatohepatitis, or primary biliary cirrhosis [71, 72]. Elevated liver enzyme levels or liver involvement was reported in 5–27% of patients with ILD and telomerase complex mutation [9, 36].
The presence of a TERT or TERC mutation increases the risk of cirrhosis developing in cases of hepatitis C infection and probably with equivalent alcohol consumption [71, 72]. Patients with the same mutation even in the same family may present normal or elevated liver enzyme levels with variable degrees of necrosis, inflammation, fibrosis and regeneration on liver histology [71].
Gorgy et al. [73] highlighted the high frequency of hepatopulmonary syndrome associated with telomerase complex mutations. In a retrospective series of 42 patients with dyspnoea and telomerase complex mutation, nine patients had minimal or absent ILD and a hepatopulmonary syndrome, defined as liver disease with evidence of intrapulmonary vascular dilatation resulting in pulmonary gas exchange abnormalities and hypoxaemia. Genetic analysis showed mutations in TERT (n=4), RTEL1 (n=1) or DKC1 (n=1), but no mutations in three patients despite their typical DKC phenotype. Among the six patients with available liver biopsies, the most common abnormality was nodular regenerative hyperplasia (n=4). The final diagnosis was non-cirrhotic portal hypertension. Two patients underwent liver transplantation and lung fibrosis developed 18 months and 12 years later [73].
Other manifestations: Other manifestations linked to telomere disease include cellular or humoral immunodeficiency, exudative retinopathies, central neurological involvement with cerebral calcifications, gastrointestinal bleeding, radiation sensitivity or infertility [31, 74, 75]. These manifestations have mainly been described in children and never or very rarely associated with adult pulmonary fibrosis [76].
Surfactant protein mutations
Surfactant is secreted by type II alveolar epithelial cells. Surfactant is composed of 90% lipids and 10% proteins [77]. Surfactant protein (SP)-A and SP-D are important for lung defence, and the hydrophobic proteins SP-B and SP-C are important for surfactant function [77]. The corresponding genes are SFTPA, SFTPB, SFTPC and SFTPD [77]. SP-B and SP-C are transported to the cell membrane in lamellar bodies by the ATP binding cassette family A, member 3 (ABCA3) transporter encoded by ABCA3 [78]. Thyroid transcription factor 1 (TITF1), a transcription factor expressed in the lung, thyroid and central nervous system, is implicated in surfactant gene transcription [79].
Among surfactant gene mutations, homozygous SFTPB mutations have been found associated with a neonatal respiratory distress syndrome but not with adult ILD [77]. SFTPD mutations have not been associated with lung disease. Among surfactant genes, SFTPC mutations are most described in adult familial ILD, but their frequency is usually <5% [23]. Transmission is autosomal dominant. Neomutations are frequent and may explain as many as 50% of cases [80].
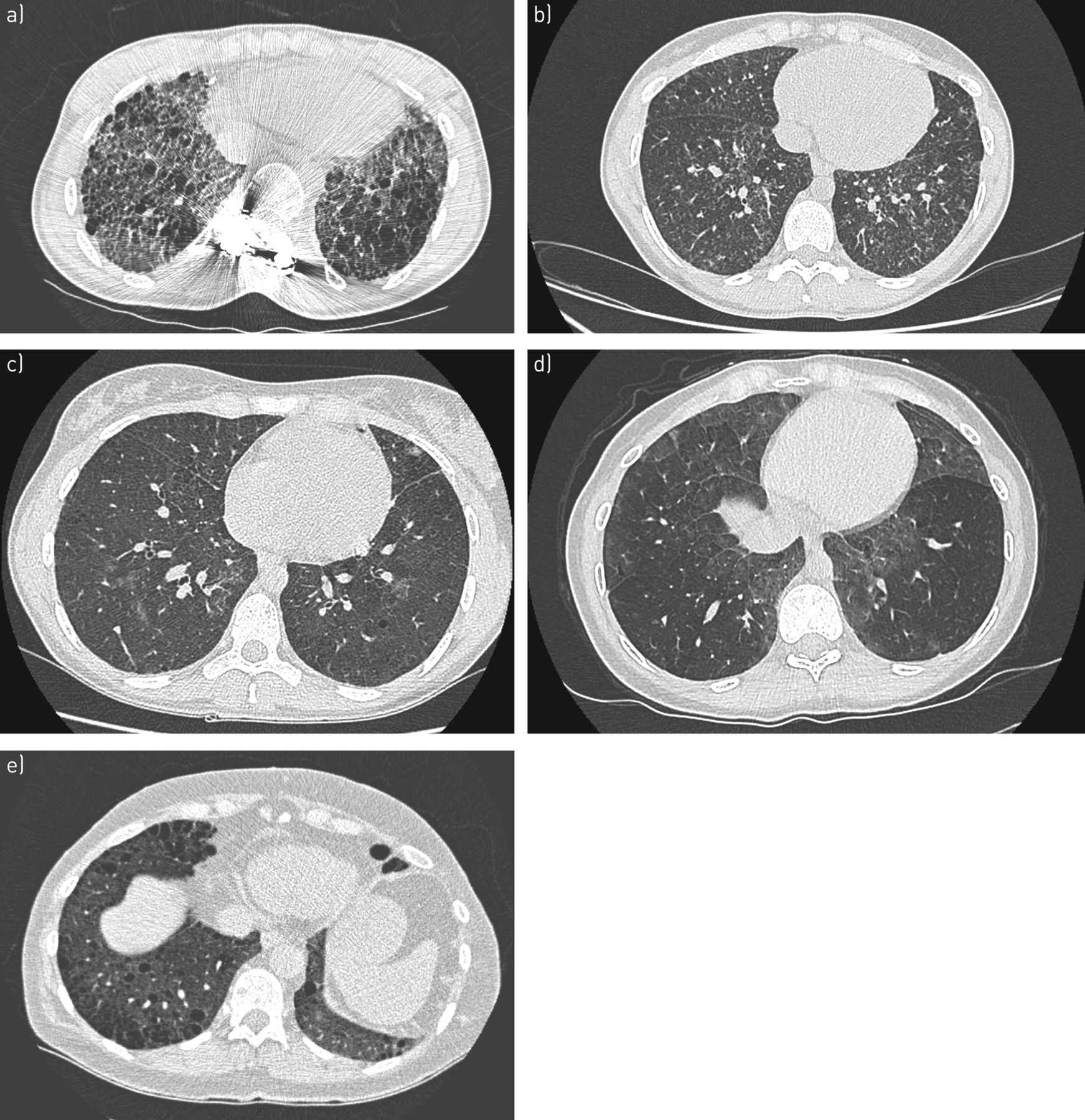
Biallelic ABCA3 mutations were first reported in children with ILD and are associated with adult ILD in cases of biallelic missense and nonsense mutations [78, 81, 82]. Biallelic ABCA3 mutations and heterozygous SFTPC mutations in adults may have a similar clinical and radiological presentation [83, 84]. The most frequent radiological pattern associates predominant diffuse ground-glass opacities, septal thickening and cysts of variable size with a preferential distribution in the upper lobes and in subpleural areas (figure 4) [85–87]. Differentiating emphysema from cysts is sometimes difficult, and SFTPC mutation must be evoked in a young patient presenting combined pulmonary fibrosis and emphysema [88]. However, at a later stage of disease, honeycombing can predominate.
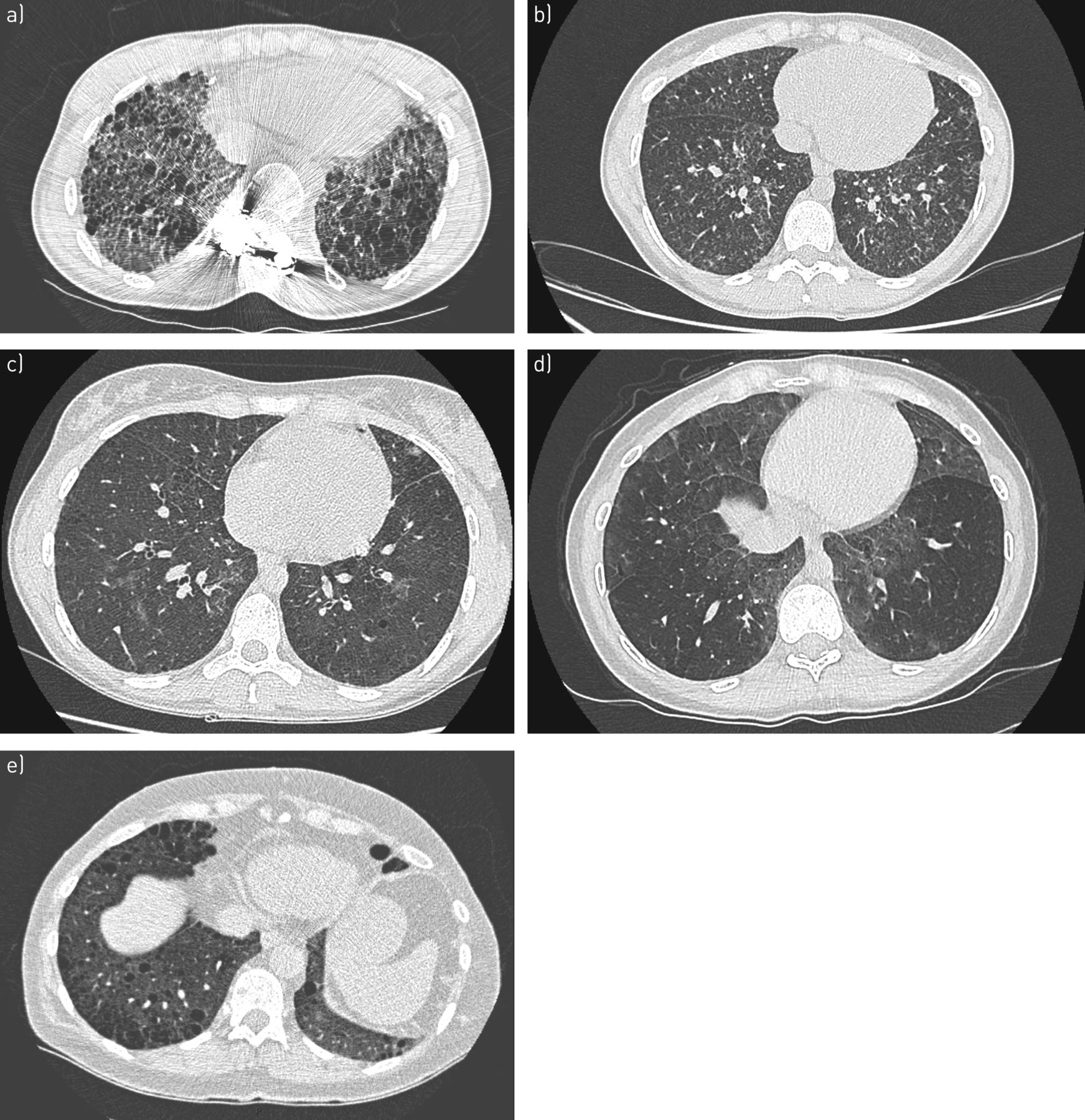
Representative computed tomography scans of carriers of a) SFTPC, b) SFTPA1, c) ABCA3, d) NFKX2-1 and e) NF1 pathogenic mutations, all inconsistent with usual interstitial pneumonia.
Histologically, the most frequently described pattern in adults is UIP, but NSIP, organising pneumonia or desquamative interstitial pneumonia have also been reported. Granulomas have not been reported, but moderate inflammation and centrolobular fibrosis can be observed [85].
In children, successful treatments reported in case reports or short series include methylprednisolone, hydroxychloroquine or azithromycin [11, 79, 86]. In any case, no treatment appears to reduce disease in a patient with predominant honeycombing lesions. The effect of antifibrotic drugs such as pirfenidone or nintedanib is unknown. The disease does not appear to recur after pulmonary transplantation [89].
Transmission of SFTPA1 and SFTPA2 mutations (figure 4) is associated with early pulmonary fibrosis and lung cancer within families. Transmission is autosomal dominant [90, 91]. In selected cases of familial ILD, study of SFTPA2, SFTPC and ABCA3 allowed a genetic cause to be identified in 6.5% of families [92].
Heterozygous NFKX2-1 mutations in children are associated with ILD, hypothyroidism and neurological anomalies (hypotonia, delayed development, chorea) [93]. This mutation may be associated with lung fibrosis in adults. The high-resolution CT pattern is usually inconsistent with UIP (figure 4) [93].
Other syndromes with pulmonary fibrosis associated with monogenic disorders
In addition to genetic disorders linked to mutations of surfactant or telomerase complex genes, pulmonary fibrosis has been described in certain rare familial syndromes with extrapulmonary involvement. Hermansky–Pudlak syndrome (HPS) associates pulmonary fibrosis, oculocutaneous albinism and platelet abnormalities and is due to mutations in the HPS-1 to HPS-9 genes [94]. Gain-of-function mutations in TMEM173 encoding stimulator of interferon genes (STING) are associated with pulmonary fibrosis and various clinical features of systemic inflammation and vasculitis involving the skin in children and young adults with a strong serum interferon elevation [7, 95]. Mutations in COPA (encoding coatomer subunit α) are associated with a syndrome of autoimmunity manifested by lung and joint disease [8]. COPA variants impair binding to proteins targeted for retrograde Golgi-to-endoplasmic reticulum transport and results in endoplasmic reticulum stress [8]. ILD and lung cystic lesions have also been reported with type 1 neurofibromatosis [96].
Each of these genes highlights a specific pathway involved in the pathophysiology of ILD. To date, and because of the rarity of these syndromes, each has to be considered individually. Specific treatments aimed at targeting the specific pathway involved may have therapeutic potential [8, 94, 97, 98]. Prevalence of the UIP pattern is low (figure 4), but pulmonary fibrosis can be at the forefront and lead to lung transplantation or death [6, 95, 99, 100].
Non-Mendelian inheritance in pulmonary fibrosis
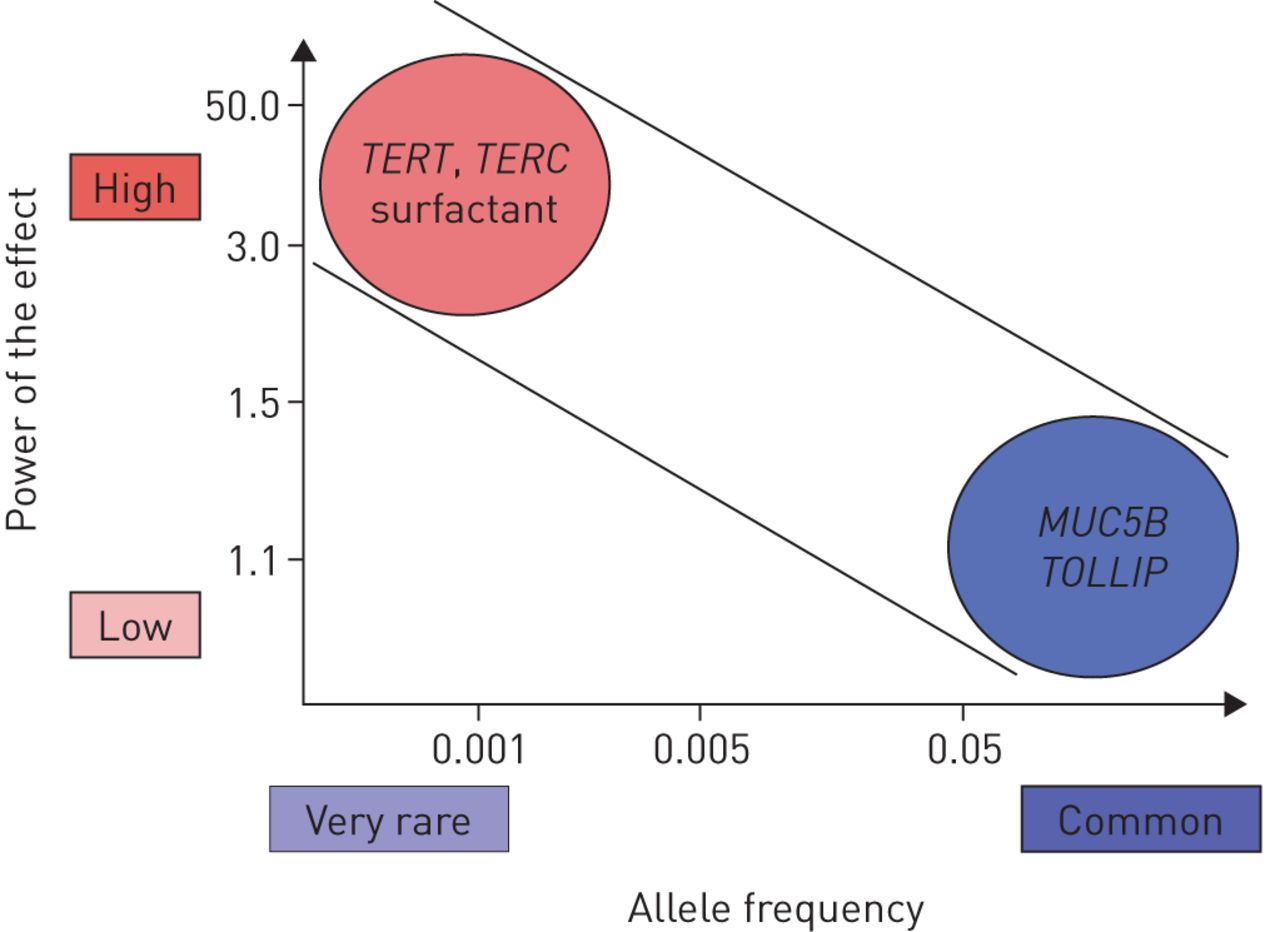
In addition to monogenic Mendelian diseases, several genetic polymorphisms increase the risk of IPF, with odds ratios up to 20 in homozygous carriers of the rare allele in the promoter of MUC5B encoding mucin-5B (figure 5) [101, 102].

Significance of the principal genetic variants associated with familial pulmonary fibrosis by their strength and frequency. For example, TERT mutations are rare but have a powerful effect with great penetrance, whereas polymorphism of the MUC5B promoter is frequent but with a weaker effect. Modified from [117, 118, 119].
Genome-wide association studies have revealed several common genetic variants associated with IPF [103, 104]: TERT (5p15 region), MUC5B (11p15 region) or the 3q26 region near TERC. The studies identified seven newly associated loci, including FAM13A (4q22 region), DSP (6p24 region), OBFC1 (10q24 region), ATP11A (13q34 region), DPP9 (19p13 region) and chromosomal regions 7q22 and 15q14-15. The results suggest that genes involved in host defence (MUC5B or TOLLIP (encoding Toll-interacting protein)), cell–cell adhesion (DSP, encoding desmoplakin) and DNA repair (TERT or TERC) contribute to the risk of fibrotic ILD.
These polymorphisms could have a clinical impact and even influence monogenic pulmonary fibrosis. Indeed, a single nucleotide polymorphism in TOLLIP, associated with mortality, was linked to reduced expression of TOLLIP, and recent post hoc analysis suggests that it affects the response to N-acetylcysteine [101, 102, 105, 106].
These polymorphisms could be considered in a polygenic transmission of pulmonary fibrosis, as MUC5B polymorphism could account for 30% of the IPF risk [107]. However, their related information cannot be used alone in genetic counselling and they are not considered in the present review.
Indication for genetic diagnosis
Genetic diagnosis may inform diagnosis and treatment
Until recently, the interest in genetic analysis has only been for enhancing knowledge, with at least two important values: 1) patients are usually eager to understand the cause of the disease and many relatives (but not all) want to know their own status (see the later section on whether genetic analysis should be made available for relatives); and 2) identifying the genetic background at the origin of the ILD can reveal a specific pathophysiological pathway and may allow for targeted therapeutic options (figure 6).
Simplified diagram of the multidisciplinary process to diagnose interstitial lung disease, including a clinician, radiologist, pathologist and also a geneticist: a first path to personalised medicine. CT: computed tomography.
From a diagnostic point of view, genetic results could be helpful and probably would limit the indication of surgical lung biopsy. Indeed, the at-risk MUC5B polymorphism is preferentially associated with the UIP pattern over other non-UIP patterns [108]. Moreover, Newton et al. [9] recently reported that the histological pattern of ILD did not influence the rate of decline of FVC in telomerase complex mutation carriers, suggesting that obtaining lung histology is useless in patient carriers of telomerase complex mutation. Some teams suggest not resorting to histological confirmation in patient carriers of telomerase complex mutation, whatever the CT scan pattern [9]. We believe that the current evidence is not yet sufficient to justify this paradigm shift, but we will have to evaluate our practices in the future.
From a therapeutic point of view, retrospective series have suggested that the genetic background could be associated with a specific response to immunosuppressive drugs [58, 59]. Indeed, carriers of telomerase complex mutations show a high rate of haematological complications after lung transplantation [58, 59] and a close evaluation of those patients is mandatory before inclusion on the transplant list, particularly when there is TERT, RTEL1, TERC, PARN, DKC1 or TINF2 mutation and thrombocytopenia <80 000 platelets·mm−3, or myelodysplastic syndrome, or somatic cytogenic abnormalities with poor prognosis.In those patients, immunosuppressive drugs are more closely monitored and tapered, and azathioprine and mycophenolate mofetil are avoided as much as possible.
Other than lung transplantation, we have no specific guidelines for treating pulmonary fibrosis associated with telomere diseases, and the usual recommendations are advised [109].
In the case of surfactant protein mutation, steroids, azithromycine or hydroxychloroquine could be effective [89]. Hydroxychloroquine is being investigated in paediatric ILD (ClinicalTrials.gov identifier NCT02615938).
Which patient is tested?
As proposed by Kropski et al. [107], we believe that genetic testing should not be offered to every patient with ILD, but instead should be limited to some specific situations.
Familial pulmonary fibrosis
Determining which patient should benefit from genetic testing lacks consensus and recommendations. International guidelines for IPF published in 2011 and 2015 did not recommend genetic testing, whereas international guidelines for idiopathic interstitial pneumonia in 2013 and French practical guidelines for IPF suggest searching for genetic abnormalities in patients with FPF [110–112]. Pending the next recommendations, there is a general agreement among experts to propose a genetic diagnosis in FPF, regardless of patient age [107].
Telomeropathy
Genetic testing should be offered to patients with pulmonary fibrosis and personal or familial telomeropathy features. With no consensual definition of telomeropathy, testing is performed according to expert opinion. We offer genetic testing in patients with ILD and haematological anomalies (unexplained macrocytosis/thrombocytopenia or myelodysplasia/aplastic anaemia), or unexplained liver disease (particularly unexplained portal hypertension) or cutaneous findings suggestive of DKC, including premature hair greying (table 2).
Patients with interstitial lung disease (ILD) for whom we propose genetic testing
Sporadic cryptogenic pulmonary fibrosis before age 50 years
Because IPF is rare before age 50 years, we offer genetic testing for sporadic cryptogenic pulmonary fibrosis occurring before age 50 years. In these cases, SFTPC and ABCA3 mutations are more common than TERT or TERC mutations, in the absence of a telomere syndrome or familial ILD [113].
Rare specific syndromes
We offer specific genetic screening for ILD with extrapulmonary manifestations such as chorea and hypothyroidism (NFKX2-1) and oculocutaneous albinism (HPS) (table 1).
Other specific situations
Genetic testing is discussed before lung transplantation [39].
Which genes to test?
Before any genetic testing, patients have to sign an informed consent that allows DNA analysis of genes associated with pulmonary fibrosis. We offer the same consultations and the same specialists for patients and relatives, including a pulmonologist with ILD expertise (either for adults or children), a psychologist and a clinical geneticist. French law distinguishes a symptomatic patient and asymptomatic relatives. All physicians can prescribe a genetic analysis to a symptomatic patient. However, asymptomatic relatives may benefit from genetic counselling.
In France, genetic analysis may be proposed by a geneticist, a genetic counsellor under the responsibility of a geneticist, or a non-geneticist physician with expertise in the clinical situation. The non-geneticist physician has to work in concert with a genetic team. Rules of good practice in constitutional genetics for medical purposes are regularly updated and should be rigorously followed [114].
The results of molecular exploration are given by the prescriber in a physical interview. Variants of unknown significance should be explained, as a familial study may help to better understand their significance.
Telomere length
As 80–90% of patients with telomerase complex mutation show reduced telomere length, some teams propose measuring telomere length before any genetic testing, and limiting genetic testing to patients with short telomeres [37, 107]. However, as 15% of patients with telomerase complex mutation present with normal telomere length, particularly in the first generation [28, 36], we do not exclude patients from genetic analysis because of normal telomere length but include telomere length, when available, in our genetic conclusion.
Gene sequencing
Methods for genetic testing are rapidly evolving and allow for several genes to be analysed together. From next-generation sequencing to whole-genome sequencing, each technique has its own advantages and disadvantages that should be specifically considered. Briefly, for patients with FPF or telomeropathy, with next-generation sequencing we propose analysing TERT, TERC, RTEL1, PARN, NAF1, DKC1, TINF2, SFTPC, SFTPA2 and ABCA3. For specific indications, we use direct sequencing to analyse genes such as TMEM173. If no mutation is identified, exome or genome sequencing could be considered.
Pitfalls of genetic analysis
One major limitation in genetic diagnosis is in the interpretation of the pathogenic role of any variant identified. Genomics guidelines and the European Society for Human Genetics have recently proposed recommendations to homogenise the genetic conclusion [115, 116]. Briefly, there are five lead categories of variants: pathogenic, likely pathogenic, variant uncertain significance (VUS), likely benign and benign. The following supporting, moderate or strong criteria are used when available: nature of the variant (very strong to supporting), previous report (strong), population data (moderate), segregation data (strong to supporting), functional data such as telomere length (supporting), computational and predictive data such as polyphen or combined annotation dependent depletion (CADD) scores (supporting) and specific phenotype (supporting). The combination of one moderate criterion and at least four supporting criteria for pathogenicity leads to a conclusion of “likely pathogenic” [115]. Some laboratories may choose to have a subclassification within the group of VUS. For instance, finding a variant leading to a stop codon within the TERT gene allows for concluding a pathogenic variant. However, a never-reported missense variant, absent from the control database, must be considered a VUS in the absence of functional and segregation data. Indeed, genetic testing for poorly selected patients will greatly increase the rate of VUS detected [107].
Genetic diagnosis and counselling in asymptomatic relatives
Genetic counselling must be performed by physicians with specific expertise. Furthermore, specific rules may apply in each country. For instance, in France, physicians are not allowed to contact relatives without the proband's agreement.
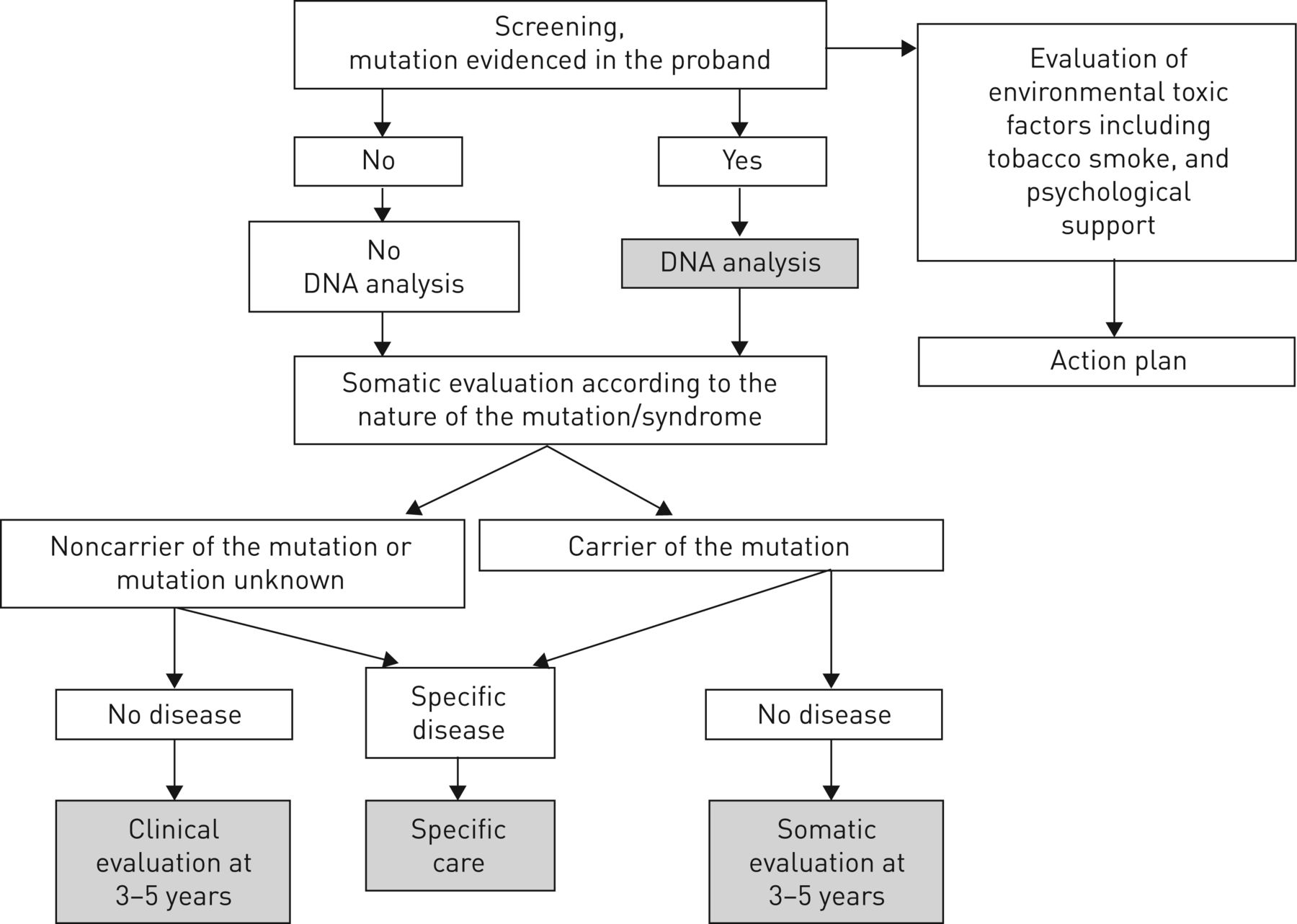
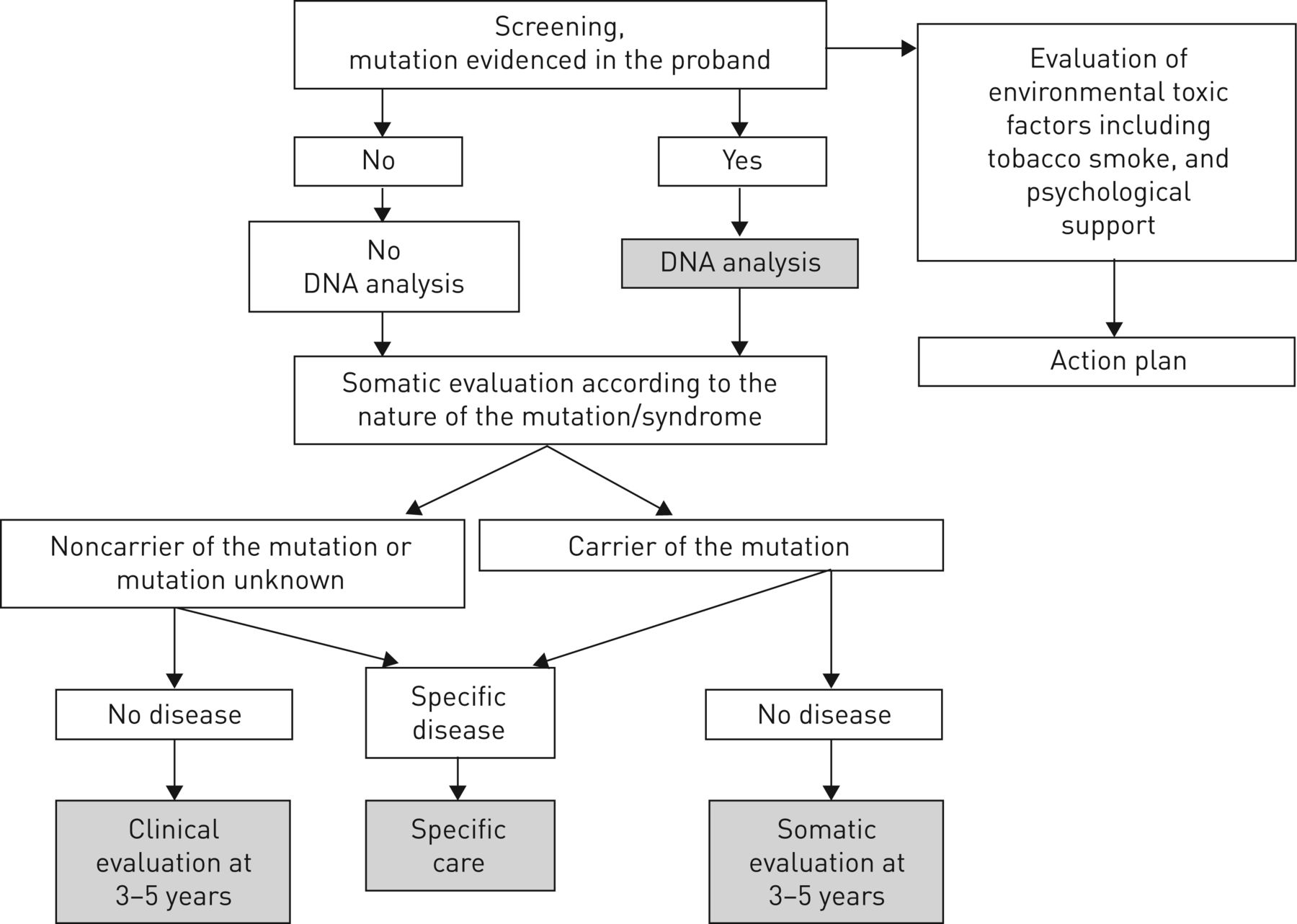
Relatives should be evaluated in a specific outpatient clinic including a pneumologist with ILD expertise, a geneticist, a haematologist and a psychologist. We always see rapidly relatives of patients with familial fibrosis who ask for specialised evaluation, because these people often have a great deal of anxiety. Genetic analysis is performed only at the request of the relatives. A blood sample for genetic analysis may be collected at the end of the first evaluation, but sampling may also be postponed according to the will of the relative (figure 7).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Diagram of genetic and somatic evaluation offered for asymptomatic relatives. Somatic evaluation includes chest computed tomography scan, pulmonary function test, complete blood count and liver blood test.
The first consultation includes an interview with a pneumologist with ILD expertise (either for adults or children), a psychologist and a clinical geneticist. According to the nature of the mutation and the possible associated phenotypes, evaluation may be scheduled with a haematologist, a rheumatologist, a dermatologist, a gastro-enterologist, a gynaecologist or a neurologist. Multidisciplinary discussion also includes at least a non-clinical geneticist, a radiologist a pathologist and a pneumologist with lung transplantation expertise.
Currently, genetic analysis takes several months. The results of genetic analysis are given and explained to the subject in our specialised outpatient clinic in a dedicated appointment scheduled 3–6 months after DNA sampling.
Should genetic analysis be made available for relatives?
If a mutation is identified in the proband, the risk for relatives can be evaluated by the type of transmission (recessive or dominant) and degree of kinship. The analysis may be specified by molecular exploration for relatives who request it. We do not offer genetic analysis to asymptomatic relatives of sporadic ILD (figure 7).
Presymptomatic genetic testing is allowed in France for relatives aged <18 years when there is evidence for a benefit. Prenatal diagnosis may be considered on a case-by-case basis.
If the mutation is not identified because of there being no DNA available for the proband, we do not perform genetic analysis for asymptomatic relatives. If no mutation is identified in the proband despite genetic analysis, the asymptomatic relative could benefit from just a clinical evaluation.
Whatever the decision regarding DNA sampling, clinicians must remember that the anticipation phenomenon in families with telomeropathy could lead to earlier and more severe disease in relatives. Indeed, we offer complete evaluation and follow-up, even in the absence of mutation in relatives of patients with telomeropathy (figure 7).
Should somatic evaluation be made available for relatives?
Whatever the presence of a mutation in the proband and the relative, we offer somatic evaluation for all relatives (figure 7). The first evaluation is based on the potential disease according to the gene involved and the pedigree. After a clinical examination with careful cutaneous, abdominal and respiratory evaluation, we may propose a complementary exam. Cutaneous examination should look at premature greying of the hair, nail dystrophy, oral leukoplakia and abnormal skin pigmentation. Abdominal examination should look at signs suggestive of portal hypertension such as swollen veins on the anterior abdominal wall and splenomegaly.
However, before prescribing evaluation to all relatives, two issues must be considered: 1) the therapeutic impact of diagnosis of asymptomatic ILD in mutation carriers is unknown; 2) the psychological impact of ILD diagnosis in relatives of patients with pulmonary fibrosis should not be minimised.
Pulmonary evaluation
Should a chest CT scan be performed?
CT scan is actually the gold standard for the screening of ILD, but the long-term impact of repeated CT scans in carriers of a mutation within genes involved in DNA repair is unknown.
In an American cohort of TERT mutation carriers, no patients younger than 40 years presented ILD, whereas the prevalence of ILD was >60% after age 60 years [28]. Systematic high-resolution chest CT in asymptomatic members of families with FPF revealed asymptomatic ILD in 14–25% of those tested [2, 16, 25]. At a median age of 50 years, 15 out of 19 asymptomatic relative carriers of TERT mutation presented opacities suggestive of ILD [66]. Age at diagnosis of ILD may be younger in patients with TERC than TERT mutations and, because of the anticipation phenomenon, patients from successive generations may present ILD at an earlier age than their parents [9].
In practice, in symptomatic patients or when clinical examination is abnormal (digital clubbing or crackles), a chest CT scan is prescribed. With asymptomatic patients without any abnormalities at clinical examination, we propose a chest CT scan at age 40 years, or 10 years before the age of onset in the proband. In the absence of ILD we propose repeating the chest CT scan after 5 years.
Should a pulmonary function test be performed?
Results of pulmonary function tests are normal in most relatives and the test should not be used as a screening tool because of its low sensitivity [2, 25]. In a series of 31 relatives with asymptomatic ILD, four subjects (12.9%) showed reduced DLCO [25]. Cardiopulmonary exercise testing also has a low sensitivity [25].
Pulmonary function tests have to be performed in symptomatic patients or when ILD is diagnosed [2, 25]. Moreover, pulmonary function tests may be considered as an initial evaluation, as further evaluation is proposed to all relatives whatever the screening of a specific disease (figure 7).
In practice, we offer a pulmonary function test as the initial somatic evaluation for all relatives. In the absence of ILD we propose repeating the pulmonary function test after 5 years.
Should a lung biopsy be performed?
In one cohort, transbronchial biopsies revealed abnormalities in 36% of asymptomatic relatives from patients with FPF, but no differences were noted in inflammatory cells on bronchoalveolar lavage between at-risk and control participants in the same cohort [2].
We do not consider transbronchial biopsies, cryobiopsies or surgical lung biopsies for asymptomatic relatives unless an ILD is detected by screening.
Should extrapulmonary evaluation be performed?
According to the nature of the mutation, the same limits exist for extrapulmonary evaluation as for pulmonary evaluation. However, routine blood tests are easy to realise and to interpret and results should probably be given by a physician with specific expertise. For instance, in a cohort of 20 asymptomatic relatives with TERT mutation, four (20%) presented macrocytosis and one (5%) thrombocytopenia, but none presented liver dysfunction [66].
Taking these results into account, a complete blood count and liver blood test are proposed for all relatives of patients with telomeropathy. Abdominal ultrasonography is added if there is an abnormal liver test.
Depending on the pedigree and the symptoms, a specialised consultation may be required with gynaecologists, hepatologists, nephrologists or rheumatologists.
Action plan
In any case, patients must be encouraged to avoid all toxic factors (respiratory, hepatic or medullar) and especially tobacco smoke, environmental toxins or cytotoxic drugs. Altered professional activities may be considered to avoid known occupational risks. Regarding the risk of anticipation and the pleiotropic effect of telomerase complex mutations, genetic counselling must be a multidisciplinary approach and discussed case by case.
Genetic multidisciplinary discussion
In Bichat hospital (Paris, France), we have developed a monthly multidisciplinary discussion involving pulmonologists specialising in ILD and lung transplantation, paediatricians specialising in ILD, geneticists, radiologists, pathologists and psychologists. Haematologists, hepatologists, dermatologists and immunologists are involved in discussions when a specific question is raised (figure 6). For each patient, this monthly multidisciplinary discussion produces three questions. 1) Molecular diagnosis: is a mutation identified and how likely is it that the mutation is pathogenic? 2) Therapeutic management: what treatment can be proposed for the lung disease (anti-fibrotic treatment, lung or medullar transplantation, others)? 3) Genetic counselling: what is the proposed advice for relatives and is there a place for a prenatal diagnosis, if asked for by the family?
Conclusions
FPF is relatively frequent and raises specific problems. Approximately 25% of families have an identified mutation and should benefit from specific genetic counselling. Current studies aim to identify new genes by whole-genome sequencing to increase this percentage and to develop specific therapies. This molecular progress comes with specific questions such as the significance of the number of VUS identified.
In adults, the most frequently detected mutations are within TERT, PARN and RTEL1. Beyond pathophysiological knowledge, detection of these mutations has practical consequence for patients. For instance, mutations in TERT and TERC are associated with haematological complications after lung transplantation and may require adapted immunosuppression, but are also associated with genetic anticipation and different phenotypes, even in the same family.
Prospective clinical trials are needed to evaluate the therapeutic options for these patients depending on their genetic background. Regarding the rarity of the disease and the limited evidence to set up specific diagnostic counselling, international collaboration is mandatory to address these crucial issues.
Disclosures
Footnotes
Support statement: This work was supported by the Chancellerie des Universités de Paris (legs Poix), la Fondation du Souffle and by a grant “FPI-SPC” from the Université Sorbonne Paris Cité and a grant from Roche. Funding information for this article has been deposited with the Crossref Funder Registry.
Conflict of interest: Disclosures can be found alongside this article at err.ersjournals.com
Provenance: Commissioned article, peer reviewed.
- Received December 23, 2016.
- Accepted February 21, 2017.
- Copyright ©ERS 2017.
ERR articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










