





Abstract
Epoprostenol was the first therapy to be approved for the treatment of pulmonary arterial hypertension (PAH). In the 20 years since the introduction of this prostacyclin analogue, the outlook for patients with PAH has improved, with survival rates now double those from the era before the development of disease-specific treatments. Today, there are a large amount of data on the clinical role of prostacyclin treatments and a body of evidence attesting the efficacy of epoprostenol in improving exercise capacity, key haemodynamic parameters and PAH symptoms, as well as in reducing mortality. The place of epoprostenol in the therapeutic management of PAH continues to evolve, with the development of new formulations and use in combination with other drug classes. In this review, we provide a historical perspective on the first 20 years of epoprostenol, a therapy that led to evidence-based study of PAH-specific treatments and the subsequent expansion of treatment options for PAH.
Abstract
The evolution of the place of epoprostenol in the management of pulmonary arterial hypertension http://ow.ly/OkY3303N2CX
Introduction
Pulmonary arterial hypertension (PAH) is a rare, progressive disease associated with significant morbidity [1–4]. The disease is characterised by elevated pulmonary artery pressure (PAP) and pulmonary vascular resistance (PVR). Left untreated, PAH leads to right-sided heart failure and premature death [1–4]. In the 1980s, median survival was 2.8 years from diagnosis; the 5-year survival rate was 34% [5]. Although PAH remains incurable, insights into the underlying mechanisms have led to the development of disease-specific treatments that have approximately doubled survival rates [6–8]. Today there are 10 approved PAH-specific therapies [9]. The first of these was the prostacyclin analogue epoprostenol, which was approved in 1995 in the USA before being licensed, a year later, in Europe. This treatment is still regarded as the gold standard to which other therapies should be compared [10–12].
In this review, we take a historical perspective on epoprostenol and its place in PAH management over the past 20 years. We also consider the role of epoprostenol at a time when new formulations that are stable at room temperature are becoming more widely available. This review is based on our knowledge of the field, supplemented by a methodical literature search designed to provide a comprehensive historical overview of milestones since the approval of epoprostenol. The literature review comprised searches of a database of PAH pdf files, PubMed, Scopus and the abstract database Searchlight for relevant publications on epoprostenol in PAH using “epoprostenol” and “pulmonary arterial hypertension” as keywords. The period for the PubMed, Scopus and Searchlight searches was 1994 to December 2014 and all English language citations were captured. Titles and abstracts of original articles were manually searched and potentially relevant articles selected; the authors reviewed the resulting publication list and agreed papers of interest. Review articles that capture the historical narrative of the development and study of epoprostenol have been included.
Background
Impact of epoprostenol on PAH treatment
Before the approval of epoprostenol, PAH was treated using a combination of non-specific treatments including warfarin, calcium-channel blockers, digoxin, diuretics and supplemental oxygen. These therapies targeted specific aspects of the disease, but demonstrated little short-term or long-term benefit on major haemodynamic parameters or clinical outcomes (reviewed in [3, 13]), with the exception of long-term calcium channel blockers that improved outcomes in a majority of patients classed using strong criteria such as acute pulmonary vasodilator responders [14]. The introduction of epoprostenol transformed the care of patients with PAH [15]: epoprostenol improved exercise capacity, key haemodynamic parameters and PAH symptoms [16, 17] and, importantly, was the first pharmacological therapy to reduce mortality [18]. Twenty years later, epoprostenol remains the only treatment to have reduced mortality in patients with idiopathic PAH (IPAH) in a randomised study [17].
Early studies of epoprostenol improved our understanding of pulmonary hypertension from associated causes, and led to evidence-based studies of PAH-specific treatments and the subsequent expansion of treatment options for PAH [19, 20]. Lessons learned from studying epoprostenol have informed the development of other inhaled, oral and subcutaneously administered prostanoid therapies. More recently, epoprostenol and some other prostacylins have been shown to be effective and well tolerated when used in combination with other PAH drug classes [21–25]. Therapies previously reserved for patients with severe disease are now being considered for use in those with earlier stage disease, in an attempt to further prolong life and improve patient outcomes [11, 12, 26, 27].
Current recommendations and evolving terminology
The 2015 European Society of Cardiology and European Respiratory Society guidelines for the diagnosis and treatment of pulmonary hypertension outline the continued place of epoprostenol within treatment options for patients with PAH (World Health Organization (WHO) group 1 pulmonary hypertension) [11, 12]. Based on level A evidence of efficacy (data derived from multiple randomised clinical trials or meta-analyses), intravenous epoprostenol is recommended as a class I monotherapy in patients with PAH (WHO group 1) with WHO functional class (FC) III or IV. It should also be considered (class IIa) for use in upfront combination therapy in patients with WHO FC III or IV alongside bosentan, and alongside bosentan and sildenafil, based on level C efficacy evidence (consensus of opinion of experts and/or small studies, retrospective studies and registries) [11, 12]. Today, epoprostenol is approved in many countries including the USA where it is indicated to improve exercise capacity in patients with WHO group 1 pulmonary hypertension (specifically, patients with IPAH, heritable PAH and PAH associated with coexisting conditions such as connective tissue disease (PAH–CTD)) based on studies including predominantly patients with New York Heart Association (NYHA) FC III–IV symptoms [28]. The term IPAH had not been developed at the time of initial approval of epoprostenol; thus, early studies describe patients with primary pulmonary hypertension (PPH) which was the preferred term at this time. Current terminology will be used within this review wherever appropriate. WHO FC and NYHA FC are used interchangeably when characterising patients with PAH.
Early research and discovery
The development of epoprostenol stemmed from the discovery of endogenous prostacyclins in the vasculature by Moncada et al. [29] in the 1970s. Soon after this, epoprostenol was synthesised and shown to have anti-platelet activity and vasodilatory effects in humans [30–32]. One of the first patients given epoprostenol was a young, cyanotic, hospitalised and bed-bound woman with IPAH. Intravenous epoprostenol improved haemodynamic parameters and clinical symptoms and the patient was discharged to continue long-term treatment [33]. Another early proof-of-concept study involving seven patients with IPAH showed that epoprostenol increased cardiac output and reduced PAP and PVR [34]. These and other early explorations preceded the innovative clinical studies that led to the first approval of epoprostenol for the treatment of patients with IPAH in 1995 [16, 17].
Epoprostenol in profile
Pharmacology
Epoprostenol is a synthetic analogue of the naturally occurring eicosanoid prostacyclin (prostaglandin I2 or PGI2), which is the main metabolite of arachidonic acid [30, 35]. Endogenous prostacyclin is produced predominantly by endothelial cells and acts both on local vasculature and on blood cells that adhere to the endothelium [26]. In PAH, the normal release of endogenous prostacyclin is depressed and release of the vasoconstrictor thromboxane A2 is increased [36]. In addition, pulmonary endothelin-1 homeostasis is abnormal, and this may contribute to the progressive rise in PVR that typifies PAH [37].
Prostacyclins (and related prostanoids) have direct and potent vasodilatory effects resulting from their action on vascular smooth muscle cells; they inhibit platelet aggregation and thrombus formation, and have antiproliferative and anti-inflammatory actions (figure 1) [26, 38]. These effects are mediated via G-protein-coupled prostanoid IP receptors in blood vessels, leukocytes and thrombocytes [26]. Epoprostenol may also have indirect vasodilatory effects owing to inhibition of production of the potent vasoconstrictor endothelin-1 [39]. In patients with PAH, therapeutic use of prostanoids is associated with immediate vasodilatory action in the pulmonary and systemic circulation and resultant, longer-term haemodynamic changes that contribute to additional decreases in PVR [26]. It has been suggested that indirect positive inotropic effects of therapy may also ameliorate systemic hypotension [26]; however, such effects have not been established in any model of chronic pulmonary hypertension, and the effect of epoprostenol in chronic pressure overload on the right ventricle remains unknown.

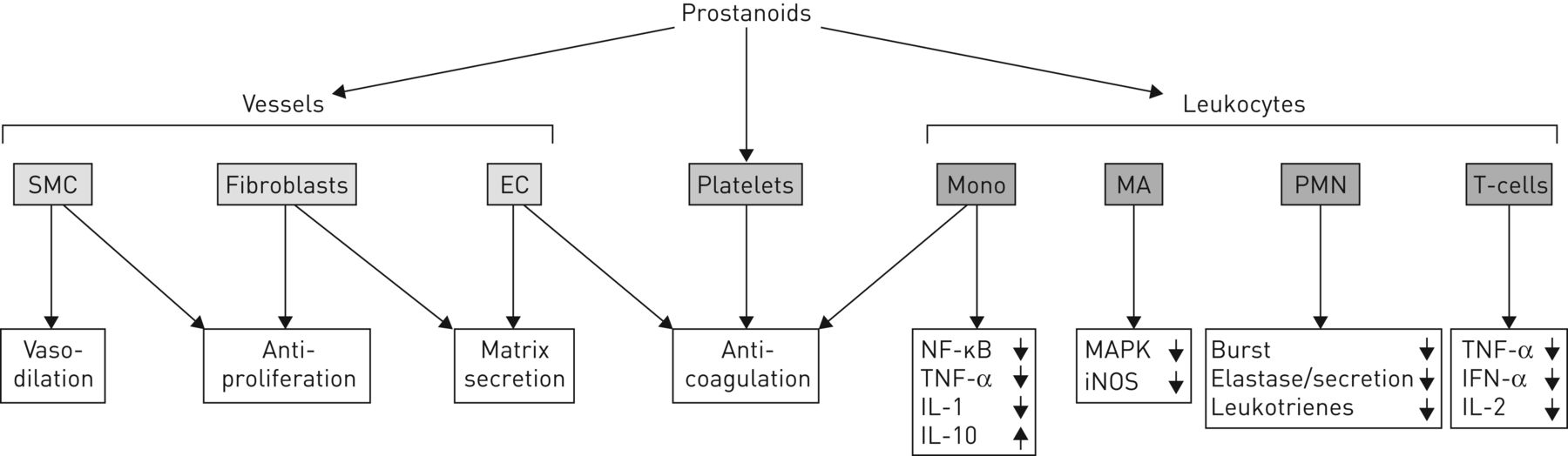{kind=link}
The effects of prostanoids on vasculature and blood cells; a variety of vascular cells, platelets and leukocytes have been identified as targets for the antiproliferative, anti-inflammatory and anti-aggregatory actions of prostaglandins. SMC: smooth muscle cells; EC: endothelial cells; Mono: mononuclear cells; NF: nuclear factor; TNF: transforming nuclear factor; IL: interleukin; MA: macrophages; MAPK: mitogen-activated protein kinase; iNOS: inducible nitric oxide synthase; PMN: polymorphonuclear neutrophils; Burst: generation of reactive oxygen species. Reproduced from [38] with permission from the publisher.
The pharmacokinetic properties of the original formulation of epoprostenol are dominated by the lability of the molecule in aqueous fluids at physiological temperature and pH. Epoprostenol has a short elimination half-life of approximately 3–6 min in human blood, which necessitates administration via continuous intravenous infusion [15]. Treatment has to be initiated by an experienced physician, and long-term use requires a permanent central venous catheter and portable infusion pump [11, 12].
Clinical studies with epoprostenol in the treatment of PAH
Today there is substantial evidence from randomised controlled trials (RCTs) supporting the use of prostacyclin treatments in PAH, while data from observational studies and registries provide real-world evidence and experiences of patient management (for reviews see [6, 40]). Table 1 provides an overview of key studies that have contributed to our understanding of the clinical profile of epoprostenol.
Overview of key studies that have contributed to our understanding of the clinical profile of epoprostenol
RCTs with epoprostenol in PAH
Epoprostenol was initially approved for use in patients with “PPH and moderate-to-severe functional status”, based on data from two RCTs [16, 17]. The first studied 24 patients with IPAH (NYHA FC II−IV), randomised to receive either intravenous epoprostenol or the conventional treatment of the time for 8 weeks [16]. Epoprostenol was associated with a significant and sustained decrease in total pulmonary resistance (–7.9 units; p=0.022) but there was no change for patients on conventional treatment (–0.2 units), and six out of 10 patients in the epoprostenol group compared with only one out of nine patients in the conventional treatment group had reductions in mean PAP (mPAP) of greater than 10 mmHg. This study also reported that continued epoprostenol treatment for up to 18 months was associated with persistent haemodynamic effects (table 1) [16]. The second, pivotal RCT was a study in 81 patients with IPAH (NYHA FC III or IV) that compared treatment with intravenous epoprostenol for 12 weeks in addition to conventional therapy with conventional therapy alone (table 1) [17]. This study demonstrated that epoprostenol treatment improved exercise capacity as shown by a median increase from baseline in 6-min walking distance (6MWD) of 31 m for patients receiving epoprostenol compared with a decrease of 29 m for those receiving conventional therapy alone (p<0.002). Key cardiopulmonary variables also improved significantly in the epoprostenol-treated group, with a change in mPAP of –8% compared with +3% in the conventional therapy group (p<0.002), and a significant mean change in PVR of –21% in those receiving epoprostenol versus +9% with conventional therapy alone (p<0.001). This study also reported that treatment with epoprostenol conferred a survival advantage over conventional therapy alone, with eight patients on conventional therapy dying during the study compared with none in the epoprostenol-treated group (p=0.003). This remains the only RCT in which a treatment approved for PAH reduced mortality.
In 2000, another pivotal RCT in patients with moderate-to-severe PAH (patients with PAH due to scleroderma (PAH–CTD)), was published (table 1) [18]. This study involved 111 patients randomised to receive continuous epoprostenol plus conventional therapy or conventional therapy alone for 12 weeks. Epoprostenol treatment was associated with significant improvements in exercise capacity at 12 weeks as demonstrated by a median change from baseline in 6MWD of +63.5 m in the epoprostenol group compared with –36.0 m in the conventional treatment only group (p<0.001). Patients receiving epoprostenol experienced improvements in haemodynamic parameters compared with those on conventional therapy alone, achieving a change from baseline in mPAP of –5.03 mmHg compared with +0.94 mmHg in the conventional therapy only group, and a mean change from baseline in PVR at 12 weeks of –4.58 mmHg·L−1·min−1 compared with +0.92 mmHg·L−1·min−1 for the conventional treatment only group. Long-term outcome data showed improved survival in patients receiving epoprostenol during a 3-year extension period compared with historical controls [44].
Non-RCTs and observational studies
There is a strong body of evidence from non-RCTs showing that long-term treatment with continuous intravenous epoprostenol is associated with sustained improvements in exercise capacity and haemodynamic parameters. Some studies also reported improved survival in epoprostenol-treated patients compared with historical controls and have supported the role of epoprostenol as a bridge to lung transplantation or heart and lung transplantation [8, 41–43, 45–49]. One of the earliest non-RCTs to describe long-term outcomes of epoprostenol therapy was an open-label, multicentre, uncontrolled study in 18 patients with IPAH (NYHA FC II−IV) [45]. This study reported that improvements in the primary endpoint, change in 6MWD from baseline at 6 months, were sustained at 18 months, and demonstrated that haemodynamic improvements, such as an increase in cardiac index and reduction in total pulmonary resistance, were maintained over 12 months of treatment. Furthermore, in the 17 patients with NYHA FC III−IV followed for 37−69 months, survival was significantly improved compared with historical controls who had not received PAH-specific therapy (p=0.045) [45]. The literature also includes reports from small-scale studies of the short- and long-term benefits of continuous intravenous epoprostenol in patients with PAH–CTD associated with scleroderma and systemic lupus erythematosus [50–53].
The survival benefits of long-term treatment with epoprostenol were also highlighted by an early US observational study that followed 69 patients with IPAH (NYHA FC III–IV) [41]. Continuous epoprostenol therapy decreased PAP and was associated with 1-, 2- and 3-year survival rates of 80%, 76% and 49%, respectively, compared with historical cohort survival rates of 56% at 20 months and 47% at 30 months [41]. Subsequently, data from two other single-centre non-RCTs also reported improved survival in IPAH cohorts treated with long-term epoprostenol. One study that followed 162 consecutive patients with IPAH reported that continuous epoprostenol treatment for at least 1 year resulted in significantly greater survival rates at 1, 2 and 3 years of 87.8%, 76.3% and 62.8%, respectively, compared with expected survival rates (based on the National Institutes of Health formula for probable survival) in the absence of disease-specific therapy (table 1) [42]. Another retrospective cohort analysis in 91 PAH patients also reported better survival for patients receiving epoprostenol than was predicted by the National Institutes of Health PPH registry survival formula [46].
A number of observational studies have explored factors predicting improved survival among patients treated with continuous epoprostenol [8, 42, 46]. For example, in a study that followed 178 patients with IPAH receiving continuous therapy, survival rates at 1, 2, 3 and 5 years were 85%, 70%, 63% and 55%, respectively [8]. Baseline variables and variables measured after 3 months on epoprostenol that were associated with poor outcome were a history of right-sided heart failure, persistence of NYHA FC III–IV on treatment, and absence of a fall in total pulmonary resistance of more than 30% relative to baseline [8].
Understanding the prognostic factors associated with response to therapy is important in PAH, because patients who are predicted to have a poor response to treatment might be candidates for early lung transplantation. By contrast, for those predicted to have a good response to treatment, it may be possible to delay decisions regarding transplantation, and indeed there is evidence that effective therapy can serve as bridge to transplantation [8].
That active treatment can both delay disease progression and improve long-term survival is well recognised today and is supported by clinical evidence. For example, in one report from a retrospective single-centre study involving 74 patients treated with first-line prostanoids, the absence of disease progression of WHO FC III at 1 and 3 years was 75% and 44%, respectively, for the 37 patients receiving epoprostenol; corresponding survival rates in these patients were 94% and 75% at 1 and 3 years, respectively [54]. Many other reports from small-cohort non-RCTs and case studies confirm the improvement in haemodynamics, exercise capacity and survival associated with continued epoprostenol treatment over a period of several years [47, 49, 55].
Furthermore, a number of recently published observational studies highlight the long-term clinical impact of continuous treatment with epoprostenol in PAH cohorts. Recent data from the French pulmonary hypertension registry provides outcomes for 209 patients with IPAH treated with epoprostenol between 2006 and 2010 [43]. After just 4 months of epoprostenol therapy, both PAH-specific treatment-naïve and PAH-specific treatment-experienced patients experienced significant improvements in 6MWD (p<0.0001 and p=0.03, respectively) and PVR (p<0.0001 and p=0.009, respectively) compared with baseline. Furthermore, NYHA FC improved in 79% of treatment-naïve and 44% of treatment-experienced patients. These benefits of epoprostenol were associated with 1- and 3-year survival estimates (from initiation of therapy) of 84% and 69%, respectively. Survival benefits were greatest in treatment-naïve patients who received upfront combination therapy with epoprostenol and oral drug(s) [43].
Studies of epoprostenol in other forms of PAH
Within the Group 1 classification of PAH, there are patients with PAH associated with clinical conditions such as CTD (as previously described), HIV infection, portal hypertension and congenital heart disease [11, 12]. Our literature review identified a number of reports over the past 20 years showing use of epoprostenol in patients with PAH associated with a wide variety of disease states. It is important to note that some of these uses are off-label in some regions and countries; prescribers are directed to their local Summary of Product Characteristics for guidance on approved indications.
An early report on the compassionate use of epoprostenol in patients with PAH secondary to other diseases, including collagen vascular disease, congenital heart disease or portopulmonary hypertension, found that all groups showed a significant reduction in mPAP and a significant increase in cardiac output compared with pretreatment values [56]. Other studies in patients with portopulmonary hypertension receiving epoprostenol showed improvements from baseline in haemodynamic parameters both pre- and post-liver transplantation. There have also been reports that some patients with portopulmonary hypertension were successfully bridged to liver transplantation through epoprostenol therapy [57–62]. Moreover, our literature search identified a case report on the use of epoprostenol in patients with Eisenmenger physiology [63]. There have been small-scale studies reporting the use of epoprostenol in patients with HIV-associated PAH: one study of six patients reported improved haemodynamic parameters and NYHA FC during acute and long-term treatment, and another study in which 20 out of 82 HIV-infected patients with PAH were given epoprostenol, reported that use of epoprostenol was associated with better survival [64, 65].
Combination of epoprostenol with other PAH-targeted therapies
Epoprostenol was initially evaluated as a monotherapy, or used in addition to the conventional treatments available before its introduction. As second-generation prostanoids and new agents targeting other PAH pathways have emerged, these have been compared with epoprostenol, and are increasingly used in combination with epoprostenol [21]. For example, a retrospective study compared first-line treatment with the endothelin-receptor antagonist bosentan in clinical trials with a historical cohort receiving epoprostenol in clinical practice [66]; no evidence was found that treatment with bosentan (followed by or with an additional treatment) adversely affected long-term outcomes compared with initial intravenous epoprostenol. The long-term efficacy of first-line epoprostenol has also been compared directly with that of first-line treatment with bosentan in 74 patients with IPAH [54]. Epoprostenol was associated with a greater improvement in exercise capacity at 1 and 3 years than was bosentan, with treatments shown to have similar effects on disease progression and survival in a matched-pairs analysis.
The Bosentan Randomised trial of Endothelin Antagonist Therapy for PAH (BREATHE)-2 study was a double-blind, placebo-controlled, 16-week assessment of epoprostenol (2 ng·kg−1·min−1 starting dose, up to 14±2 ng·kg−1·min−1) alone or in combination with bosentan (62.5 mg twice daily for 4 weeks, then 125 mg twice daily) in 33 patients with PAH [67]. There was a trend towards greater reductions in the primary endpoint for the combination of epoprostenol plus bosentan compared with epoprostenol monotherapy; combination therapy was associated with a 36.3% reduction in total pulmonary resistance compared with a 22.6% reduction in the monotherapy group (p=0.08). More recent data from an observational study reported a significantly greater improvement in PVR after 4 months with the combination of bosentan and epoprostenol compared with epoprostenol monotherapy (p=0.0001) [22]. In this retrospective study of 23 patients with PAH (NYHA FC III–IV) treated with a first-line combination of epoprostenol plus bosentan, the 1-, 2-, 3- and 4-year overall survival estimates were 100%, 94%, 94% and 74%, respectively; transplant-free survival estimates at these same time points were 96%, 85%, 77% and 60%, respectively. The authors reported that, compared with matched controls, there was a trend towards an overall survival benefit for patients receiving combination therapy (p=0.07) [22].
Epoprostenol has also been studied in combination with phosphodiesterase-5 inhibitors. The addition of sildenafil (20 mg three-times daily titrated to 80 mg three-times daily) to long-term epoprostenol was evaluated in a 16-week, double-blind, placebo-controlled RCT in 267 patients with PAH [23]. Results showed a placebo-adjusted increase from baseline of 28.8 m in 6MWD in the combination treatment group (p<0.001). By week 16, combination therapy resulted in a longer time to clinical worsening than epoprostenol alone (p=0.002) [23]. Results from a small-scale observational study suggested sustained improvements in haemodynamic parameters and exercise capacity in three patients with PAH on long-term epoprostenol given add-on sildenafil [24]. A pilot study has also investigated epoprostenol in combination with tadalafil in four patients, suggesting combination may be beneficial in patients with advanced disease [68]. There are also reports of combinations of two prostanoids, such as a small-scale non-RCT report combining intravenous epoprostenol with inhaled iloprost in patients with poor tolerability of intravenous therapy alone. This eight-patient study reported that combination of these prostanoids improved mPAP, cardiac index, mixed venous oxygen saturation and systemic arterial oxygen pressure compared with epoprostenol alone [69].
One of the latest reports of combination therapy with epoprostenol comes from retrospective registry data and describes combination of epoprostenol, bosentan and sildenafil [25]. In 18 of 19 newly diagnosed patients with severe idiopathic or heritable PAH in NYHA FC III–IV, initial triple combination therapy for 4 months was associated with major improvements from baseline in 6MWD and haemodynamics with a fall in mean PVR of approximately 67% after 4 months of combination therapy. NYHA FC dramatically improved from baseline with only one patient remaining in FC III after 4 months. Improvements were sustained in the long-term in all 18 patients. One patient did not improve on therapy and underwent lung transplantation at month 3. Transplant-free survival estimates were 94% at 1, 2 and 3 years.
There has also been an observational study of the efficacy of adding prostanoid therapy, including epoprostenol, in patients with PAH who were deteriorating on first-line oral bosentan or on bosentan and sildenafil [70]. In a study involving 16 patients requiring add-on prostanoids, six patients deteriorating on oral treatment received epoprostenol. Adding epoprostenol improved 6MWD by 83 m, with the addition of a prostanoid therapy resulting in significant improvements in 6MWD after 4 months (p<0.001) and significant improvements in NYHA FC (p=0.002). These findings were similar to those reported in the recent French pulmonary hypertension registry study described earlier [43], which showed benefits to be greater in patients receiving epoprostenol as first-line therapy (either alone or particularly in combination) than in those with previous exposure to other PAH-specific treatments (94% on oral therapy/therapies). Thus, patients responding poorly to first-line oral therapies may have benefitted from earlier initiation of epoprostenol or upfront combination therapy.
The results of these studies highlight that, more than 20 years after its first authorisation, epoprostenol continues to be important in the therapeutic management of patients with PAH.
Safety profile
The safety profile of epoprostenol in PAH has been well characterised in RCTs and during real-world clinical practice [8, 16–18, 28, 41–43, 45–47, 49, 54, 55].
Treatment is associated with dose-related adverse events during initiation and dose escalation, the most common of which are generally related to the vasodilatory effects of the agent. The most common epoprostenol dose-limiting events include nausea, vomiting, headache, hypotension and flushing, in addition to chest pain, anxiety, dizziness, bradycardia, dyspnoea, abdominal pain, musculoskeletal pain and tachycardia. adverse events associated with chronic administration of epoprostenol include hypotension, bradycardia, tachycardia, bleeding, thrombocytopenia, headache, jaw pain, flushing, influenza-like symptoms, diarrhoea, nausea and vomiting. Other adverse events, including infection and thromboembolic events, are typically related to the central venous catheter drug-delivery route [15, 28].
In the RCTs described, the most common adverse events were jaw pain, diarrhoea, headache, flushing, anorexia, photosensitivity, nausea and vomiting, and reported serious complications included catheter-related sepsis, cellulitis, haemorrhage, pneumothorax and thrombosis (see table 1 for details from key trials) [16–18]. During controlled PAH trials of up to 12 weeks' duration, the local infection rate was about 18%, and during long-term follow-up sepsis was reported at a rate of 0.3 infections per patient per year in patients treated with catheter-infused epoprostenol [15, 28].
Some of the adverse events associated with treatment administration, such as catheter-related infection, can be minimised through adherence to standard practices of safe treatment administration, such as protocols for sterile drug preparation and clinical guidelines and local practice protocols for intravenous drug administration [71–73].
Recent perspectives: reformulations
The continued importance of epoprostenol within the PAH armamentarium is highlighted by the development of new formulations. The original epoprostenol formulation is unstable at physiological pH and temperatures, and must be reconstituted with a specific sterile diluent. After reconstitution, the solution must be kept cold and administered within 8−12 h if stored at room temperature, or within 24 h if maintained at 2−8°C using cold gel packs. Any reconstituted product must be discarded after 48 h [28, 74]. Accordingly, epoprostenol has to be freshly prepared, and medication cassettes changed every 12 h (or every 24 h if using cold gel packs), which can be inconvenient for the healthcare team and for patients [74].
In an attempt to overcome some of these handling limitations, new formulations of epoprostenol have been developed in recent years [75–78]. These include Veletri (epoprostenol AS; Actelion Pharmaceuticals, Allschwil, Switzerland), which is a formulation of epoprostenol containing the excipients arginine and sucrose; these improve the stability of epoprostenol in solution [79]. This formulation is reconstituted using sterile water for injection or sterile saline (sodium chloride 0.9%) and has a pH that increases with the concentration of the solution, ranging from 10.8 to 11.9 [76, 79]. When freshly prepared, Veletri is stable at room temperature (25°C) for 48 h at concentrations of 3000–<60 000 ng·mL−1and for 72 h at concentrations ≥60 000 ng·mL−1. Once prepared, it may be refrigerated (at 2−8°C) for up to 8 days; the solution is stable at room temperature for 24−72 h (depending on final concentration) after removal from refrigerated storage [76, 79]. There is also a newly developed formulation of Flolan (epoprostenol sodium; GlaxoSmithKline, NC, USA) that is reconstituted using a sterile diluent with a higher pH than the previous formulation (12.0 versus 10.5); this improves the stability of epoprostenol sodium in solution. The reconstituted product is stable at room temperature (25°C) for up to 72 h (either freshly prepared or following refrigeration for ≤8 days at 2−8°C). This newly developed formulation of Flolan (either freshly prepared or following refrigeration for ≤8 days), is also stable at 35°C for 24 h [80].
Several recently published studies have described the impact of transitioning patients with PAH from stable, conventional intravenous epoprostenol treatment to the newer epoprostenol formulations. Epoprostenol AS has been evaluated in two single-arm, open-label studies: Epoprostenol for injection in pulmonary arterial hypertension (EPITOME)-2 and EPITOME-4 [74, 81]. EPITOME-2 was a multicentre European and Canadian study that assessed outcomes in 41 clinically stable patients with PAH, followed for 3 months after transition from continuous intravenous epoprostenol to epoprostenol AS. The study reported no significant changes from baseline in any efficacy endpoints (6MWD, NYHA FC or haemodynamics), and adverse events were consistent with those previously described for intravenous prostacyclin therapy. The authors reported an improvement from baseline to 3 months in patient-reported satisfaction with treatment, in the domain of treatment convenience, as assessed using Treatment Satisfaction Questionnaire for Medication scores [74]. EPITOME-4, a two-site Japanese study, reported similar outcomes, finding that patients with PAH could be transitioned to the new formulation, to receive the same therapeutic dose of epoprostenol with no change in efficacy or safety profile [81]. Most recently, observational data have been published that highlight the equivalence of conventional and newer formulations of epoprostenol. Prospective data from the US PROSPECT registry describes the course of PAH in patients treated with the new formulation of room-temperature-stable epoprostenol [82]. This study reported a 1-year freedom from hospitalisation rate of 51% and overall 1-year survival estimate of 84% in 336 patients with PAH receiving the new formulation.
The efficacy of the pH-adjusted reformulation of epoprostenol has also been evaluated in a multicentre, single-arm study [80]. This open-label study assessed outcomes in 16 patients with PAH, transitioned to 4 weeks of treatment with the reformulated product following a 4-week run-in period on conventional epoprostenol therapy. Patients had been on a stable dose of epoprostenol for at least 3 months prior to the study. They were transitioned to the new formulation with no changes in therapeutic dose or in efficacy or safety profile. There were no significant changes from baseline (i.e. before the transition to the reformulated product) in the dose of epoprostenol, Short-Form 36 scores, 6MWD, Borg dyspnoea index, WHO FC or mean N-terminal pro-B-type natriuretic peptide levels. The number and percentage of patients with adverse events considered to be related to study drug by the investigator (one patient and three patients in the run-in and treatment periods, respectively) or who had serious adverse events (two patients and one patient, respectively) were low and similar in the run-in and treatment periods. None of the serious adverse events were considered related to the study drug. There were small improvements in mean scores on the majority of 15 items in a study-specific questionnaire about health-related quality of life, and 14 of the 16 patients (88%) preferred the reformulated product to the previous formulation [80].
Conclusion
Epoprostenol has been an important treatment for PAH for two decades and its place in patient management continues to evolve. As the first disease-specific treatment for PAH, and the first therapy shown in an RCT to improve patient survival, epoprostenol transformed PAH management and opened the door to a new era of scientific and clinical study of PAH. With a wealth of clinical data supporting the efficacy and tolerability profile of epoprostenol, this agent remains a key treatment option in PAH, and the development of new, more convenient formulations of this therapy looks set to ensure a continuing role for epoprostenol within the management of patients with PAH.
Disclosures
Supplementary Material
A. Vonk_Noordegraaf ERR-0055-2016_Vonk_Noordegraaf
O. Sitbon ERR-0055-2016_Sitbon
Acknowledgements
Winnie McFadzean and Harriet Crofts (PharmaGenesis, London, UK) provided medical writing support funded by GlaxoSmithKline (Brentford, UK). The views expressed in this review are those of the authors. Both authors contributed to the conception of the manuscript, the review of literature, and the writing and review of the article.
Footnotes
Conflict of interest: Disclosures can be found alongside this article at err.ersjournals.com
Provenance: Submitted article, peer reviewed.
- Received June 2, 2016.
- Accepted August 28, 2016.
- Copyright ©ERS 2017.
ERR articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










