





Abstract
Sarcoidosis is a systemic disease of unknown cause with very diverse presentation, outcome, severity and need for treatments. While some presentations may be very typical, for many patients, the presentation is nonspecific, with shared associations with other diseases at times being by far more frequent or misleading, which can be a cause of significant delay and often several consultations before a diagnosis of sarcoidosis can be confirmed. This is particularly the case when pulmonary manifestations are in the forefront. The diagnosis relies on three well-known criteria. In clinical practice, these criteria are not easily implemented, particularly by physicians without expertise in sarcoidosis, which can lead to a risk of either under- or over-diagnosis. Qualifying the presentation according to sarcoidosis diagnosis is essential. However, it is often not easy to classify the presentation as typical versus compatible or compatible versus inconsistent. Further investigations are needed before any other hypothesis is to be considered. It is important to detect events and to determine whether or not they are indicative of a flare of sarcoidosis. Eventually, treatment needs to be related to the correct indications. The evaluation of the efficacy and safety of treatments is crucial. To address such issues, we present five emblematic cases that illustrate this.
Abstract
Showing emblematic cases is the best way to illustrate the multiple conditions to deal with for managing sarcoidosis http://ow.ly/10A5Yv
Introduction
Sarcoidosis is a systemic granulomatous disease of unknown cause characterised by highly diverse presentations and by the absence of any single criterion allowing a definitive diagnosis [1–3]. Moreover, sarcoidosis may also have potential differences according to outcome, severity and need for therapy [2, 4, 5]. Despite the fact that some presentations may strongly suggest sarcoidosis (e.g. Löfgren's syndrome) [6], typical skin or ocular manifestations can lead to a rapid diagnosis for at least one third of patient symptoms. This may often be nonspecific and have shared associations with more frequent diseases, resulting in a diagnosis that is delayed by months to years [7]. Occasionally, rare misleading manifestations, although within the realm of sarcoidosis manifestations, can lead to sarcoidosis being missed among the gamut of possible diagnoses by physicians inexperienced in recognising sarcoidosis, particularly in elder patients [8–14]. Other diagnosis problems may arise during outcome, i.e. are new manifestations of a visceral localisation specific to sarcoidosis or to comorbidities, weighing a significant impact on therapeutic decisions? Another difficulty may arise from lung manifestations worsening: does it reveal a true disease flare or another cause? This may be difficult to answer and thereby calls for an appropriate evaluation and discussion [15, 16]. Assessing the outcome (worsening or not) or response to any treatment (or no response) may also possibly be difficult, yet it is important for deciding how to plan further treatment.
The diagnostic criteria of sarcoidosis are well known but can be difficult to apply in everyday life: it may occasionally be difficult to determine whether a presentation is compatible or inconsistent. Furthermore, decisions for therapy often rely on criteria such as progression, which deserves discussion; recently, very interesting papers have been published on this topic [15, 16].
Thus, we present five emblematic cases to illustrate certain commonly encountered situations in clinical practice that raise questions regarding an initial positive diagnosis of sarcoidosis, in addition to diagnosis of events during follow-up and treatment.
Case 1
A 52-year-old Caucasian woman was investigated due to the fortuitous discovery of a right voluminous laterotracheal lymphadenopathy on chest radiography in 1992, in the context of a non-insulin dependent diabetes mellitus and arterial hypertension. There was no indication of any other visceral involvement besides the detection of a significant increase (by 80%) in CD4+ lymphocytes (to 51%) in the bronchoalveolar lavage. Noncaseous granulomas were found by mediastinoscopy and multiple tests for Mycobacterium tuberculosis were negative. First, a diagnosis of sarcoidosis was considered. No treatment was required and a follow-up appointment was scheduled. Moreover, due to the persistence of the voluminous mediastinal lymphadenopathy, the patient was later referred to a tertiary sarcoidosis centre in 2012. The only events between 1992 and 2012 were coronary arterial disease treated by a quadruple coronary bypass, a retinal microangiopathy and a need for insulin. The patient was in good condition with a normal physical examination. Serum biological tests including angiotensin converting enzyme (ACE) were normal except that an increased glucose level and a low γ-globulin level were observed. Serum IgG concentration was 0.32 g·dL−1, IgA 0.04 g·dL−1 and IgM 0.011 g·dL−1. High-resolution computed tomography (HRCT) disclosed voluminous paratracheal and inter-tracheobronchial lymph nodes in the absence of hilar lymph node and pulmonary infiltration (figure 1). Interestingly, the patient was not prone to infections. Tests for autoantibodies were negative. A final diagnosis of common variable immune deficiency-associated granulomatosis was then considered and sarcoidosis was eventually excluded.
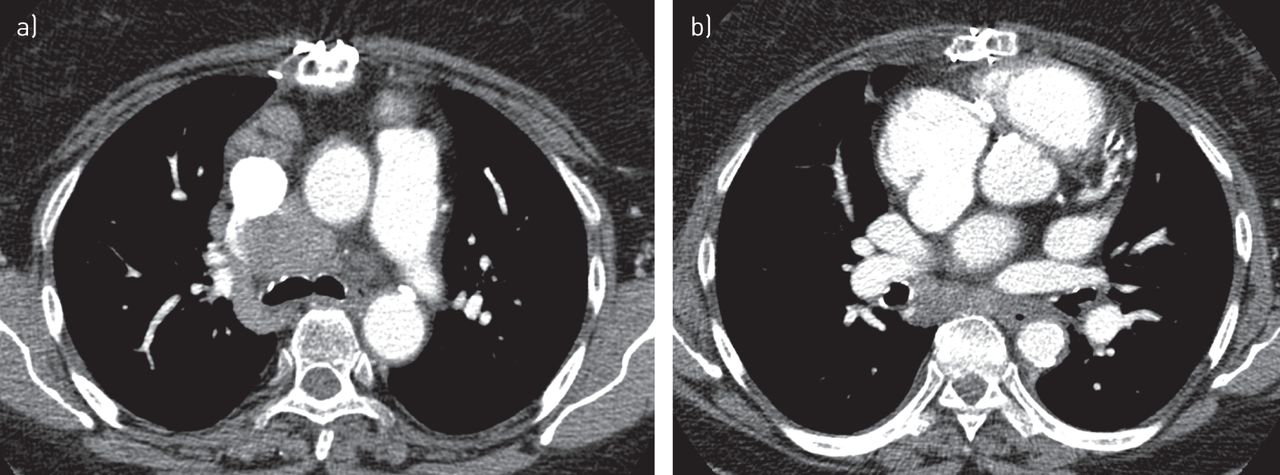
Thoracic high-resolution computed tomography: a) mediastinal lymphadenopathy b) without hilar lymphadenopathy (case 1).
Case 2
A 48-year-old Caucasian male was referred to our department for progressively worsening exertional dyspnoea, dry cough and chest pain, which had started 4 months prior. The patient had a history of gastro-oesophageal reflux treated by esomeprazole and was also a former smoker with a 20-pack-year history. He worked as a subway operator in Paris, France. In 2005, he was exposed to a significant amount of dust due to taking up home renovations. On physical examination, the patient was noted to be in good condition, showing signs of bibasilar inspiratory crackles on chest auscultation without squeaks, digital clubbing or extrapulmonary symptoms. The chest radiography showed bilateral hilar lymphadenopathy with diffuse pulmonary infiltration. The thoracic HRCT revealed enlarged bilateral hilar and mediastinal lymph nodes, with predominant diffuse ground-glass opacities and a lymphatic distribution of micronodules with fissural spreading (figure 2). The ECG, Holter monitor and echocardiogram were normal, as well as the ophthalmological examination. The patient's blood cell count, liver and renal function tests, serum calcium, and plasma protein electrophoresis did not disclose any abnormality except lymphopenia (0.94×109 lymphocytes per L). A serological test for HIV was negative. His serum level of ACE was elevated to 160 U·L−1 (normal <41 U·L−1). Biopsy of a minor salivary gland was not informative. The patient underwent a flexible bronchoscopy and the bronchoalveolar lavage showed a total cell count of 140 000 cells·mL−1, with 52% lymphocytes, 40% macrophages, 5% neutrophils and 1% eosinophils. The CD4/CD8 T-lymphocyte ratio was 4.2. Endobronchial mucosal biopsies showed nonspecific inflammation and transbronchial lung biopsies revealed non-necrotising epithelioid granulomas. Pulmonary function tests revealed a restrictive pattern with: total lung capacity 4.61 L, corresponding to 71% of the predicted value; forced vital capacity (FVC) 2.39 L, corresponding to 65% of the predicted value; and reduced diffusing capacity of the lung for carbon monoxide (DLCO) corresponding to 62% of the predicted value. The 6-min walk distance was 595 m, without oxygen desaturation. Altogether, these findings allowed the diagnosis of sarcoidosis to be confirmed.
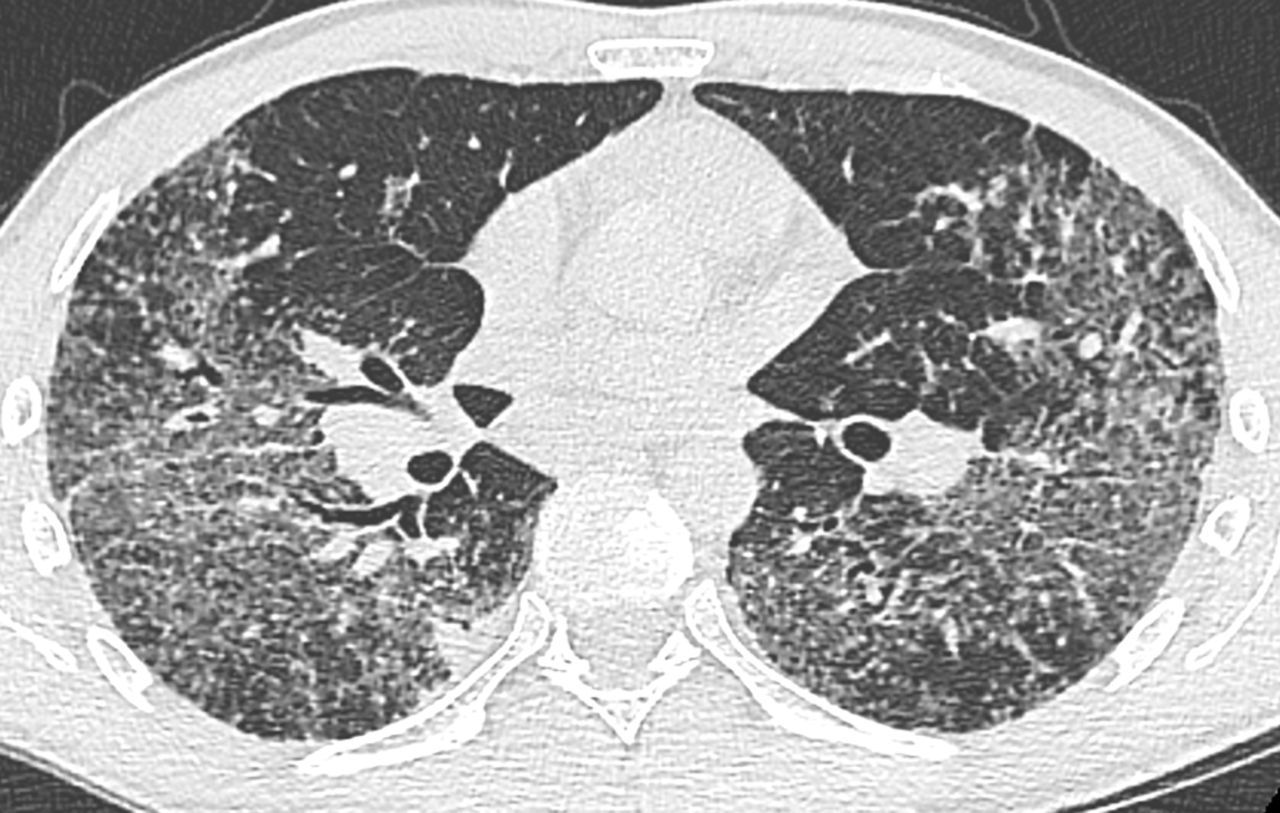
Thoracic high-resolution computed tomography: predominant diffuse ground-glass opacities associated with a lymphatic distribution of micronodules with fissural spreading and bilateral hilar lymphadenopathy (case 2).
Case 3
A nonsmoking, 34-year-old woman was diagnosed with sarcoidosis in 2011. Both her parents were originally from the West Indies. Her medical history was notable for pernicious anaemia and gastritis, thereby receiving monthly intramuscular B12 injections. Clinical manifestations started with a persistent cough. Chest radiography showed bilateral hilar lymphadenopathy and diffuse pulmonary micronodules. Thoracic HRCT lesions were typical of sarcoidosis. Serum ACE level was three times the upper limit of the normal range. Histological noncaseating epithelioid granulomas were observed on endobronchial biopsies, associated with lymphocytosis on bronchoalveolar lavage with CD4/CD8 ratio at 6.46. A liver biopsy was performed because of asymptomatic moderate cholestasis. Pathological analysis confirmed sarcoidosis localisation. A diagnosis of stage 2 thoracic sarcoidosis with liver involvement was made. On July 2011, the patient started receiving prednisone 40 mg per day (2/3 mg·kg−1). 2 months later, she experienced syncope and sarcoidosis-specific localisation could not be ruled out from the cardiac magnetic resonance imaging. Resting ECG, echocardiography and Holter monitor results were normal, and the probability of a cardiac sarcoidosis was considered intermediate. In this case of multivisceral sarcoidosis and due to 15 kg weight gain from steroid therapy, azathioprine (150 mg per day) was started in May 2012. A favourable outcome allowed for the reduction of treatment; later, azathioprine was discontinued on September 2013. In April 2015, she was on prednisone 8 mg per day for 6 months, when she was admitted to an emergency department for fatigue, tachycardia, acute abdominal pain and diarrhoea. Both abdominal ultrasound scan and chest radiography were normal. Blood tests revealed an increase in the total serum calcium level to 2.88 mmol·L−1 (normal range 2.15–2.50 mmol·L−1) and 3.08 mmol·L−1 for albumin-corrected serum calcium levels. The patient was referred to an internal medicine department, and a physical examination showed a thyroid goitre. Clinical symptoms and plasma calcium levels improved under intravenous infusion of physiological saline solution. The diagnostic evaluation performed for the understanding of hypercalcaemia is summarised in table 1. The main results included: normal serum protein electrophoresis, no liver cholestasis, moderate increase of ACE levels (51 IU·L−1, upper limit 41 IU·L−1), no hypercalciuria (0.001 mmol·kg−1 per 24 h), low parathyroid hormone plasma levels (9.4 ng·L−1, normal range 15–65 ng·L−1), normal lymphocyte count (1920 per mm3), lower than normal 25-hydroxy- and 1,25-dihydroxyvitamin D3 plasma levels. Additionally, thyroid-stimulating hormone (TSH) plasma levels were undetectable and thyroxine plasma levels were extremely high at 100 pmol·L−1. Notably, TSH plasma level was normal 4 months prior. Therefore, thyrotoxicosis was a rational explanation for the patient's clinical symptoms at admission. Autoimmune thyrotoxicosis was confirmed by the presence of antibodies to the TSH receptor and thyroid peroxidase antibodies in this patient, who was already known to have pernicious anaemia. There was no evidence of sarcoidosis activity that could cause hypercalcaemia; according to the results, hypercalcemia was related to Graves' disease. Carbimazole treatment was started with no change in her daily prednisone dose. Serum calcium level normalised during the first month of treatment. In this case, after a diagnosis of sarcoidosis had been confirmed, the occurrence of a significant hypercalcaemia led to consideration of Graves' disease as the origin of this metabolic event, in which sarcoidosis was not involved.
Case 3
Case 4
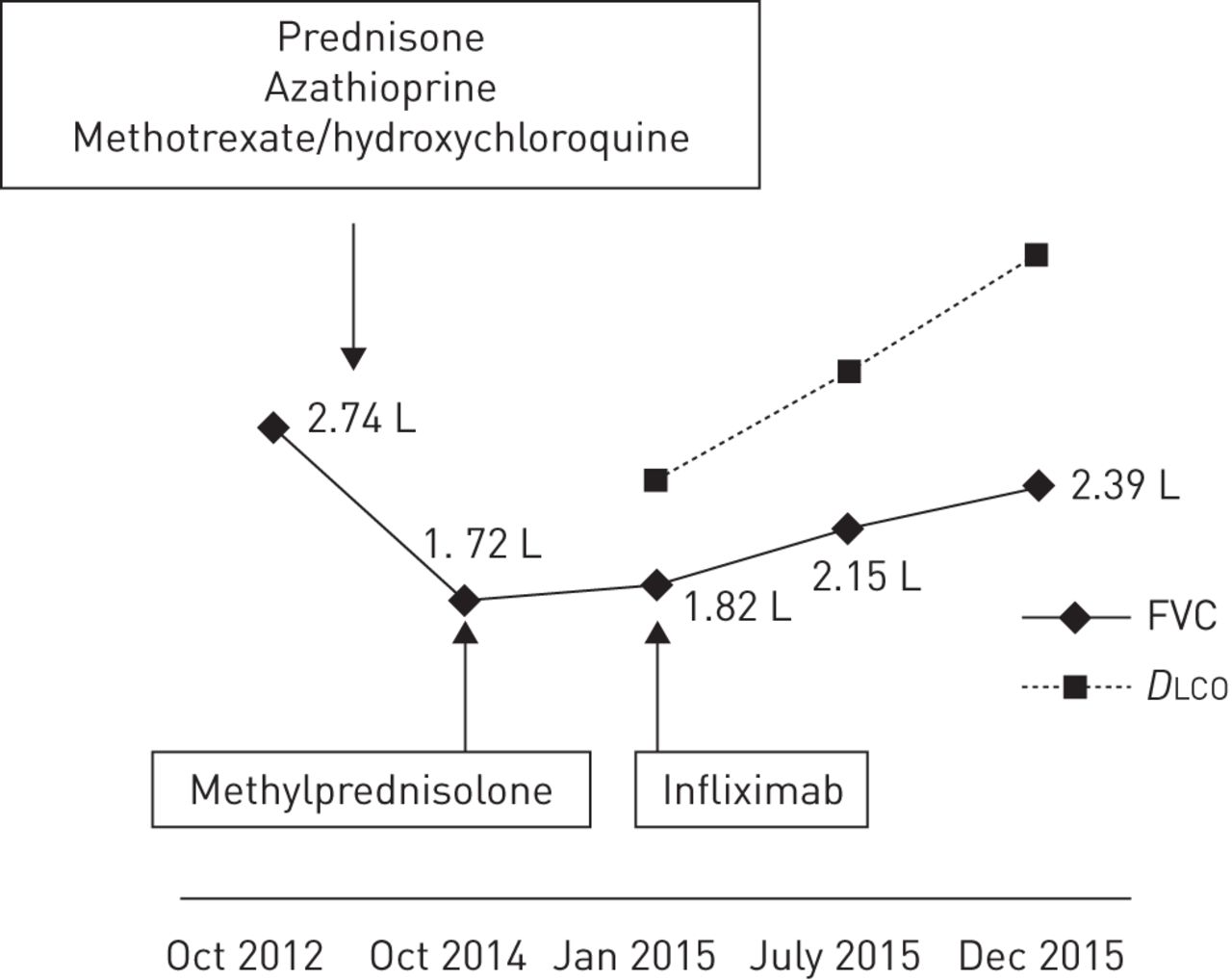
In January 2012, a 41-year-old woman was admitted to our outpatient clinic for 1-year-old symptoms of cough, wheezing, frequent insomnia, nocturnal hyperhidrosis and fatigue. Due to a history of asthma during childhood, inhaled salbutamol treatment was given without any improvement. The patient was born in the French West Indies and worked as a homecare assistant. She had no history of drug, food or seasonal allergy. She was, however, an active smoker using a half a pack of cigarettes per day, and collectively having a 15-pack-year smoking history. Physical examination showed New York Heart Association (NYHA) functional class II dyspnoea, normal cardiopulmonary auscultation, no peripheral lymph nodes, and neither hepatomegaly nor splenomegaly. Chest radiography uncovered bilateral hilar lymphadenopathy and bilateral pulmonary infiltrates (stage 2) (figure 3a). Thoracic HRCT confirmed bilateral hilar and mediastinal lymphadenopathy, identifying typical perilymphatic micronodules and a few areas of consolidation. There were no signs of parenchymal fibrosis. There was no evidence of extrathoracic manifestations and the resting ECG was normal. On flexible bronchoscopy, the bronchial tree had a normal appearance while the bronchial mucosal biopsies showed noncaseating granulomas. Bronchoalveolar lavage cell count was 494 000 cells·mm−3 with 54% pigmented macrophages, 42% lymphocytes (CD4/CD8 ratio 2.26) and 4% neutrophils. Serum calcium level was normal. C-reactive protein level was mildly increased at 22 mg·L−1. Blood lymphocyte count was normal (1448 lymphocytes per mm3). Serum ACE level was three times the upper limit of the normal range. Pulmonary function tests at baseline were abnormal with FVC 2.57 L (66% pred) and forced expiratory volume in 1 s (FEV1) 2.11 L (66% pred) with no significant post-bronchodilator improvement (+160 mL, 8%) (figure 4). DLCO was not measured. Prednisone treatment started in February 2012 at 0.5 mg·kg−1 prednisone per kg, i.e. 30 mg per day. The patient experienced a significant weight gain from 62 to 81 kg. Thus, azathioprine was introduced as a steroid-sparing agent in July 2012. Finally, the patient stopped prednisone on her own, 2 months later, while continuing with azathioprine alone. In February 2013, there was no clinical improvement observed with persistent radiological and biological activity. Methotrexate (10 mg orally twice a week) and hydroxychloroquine (400 mg·day−1) replaced azathioprine. This treatment was effective, and 4 months later, clinical, radiographic and ACE plasma level improvement were observed. However, hepatic blood tests showed increased aspartate and alanine aminotransferase, to five times the upper limit of the normal range, while the patient additionally was taking paracetamol. Liver blood tests normalised after discontinuation of all drugs. Methotrexate at a lower dose (10 mg per week) and hydroxychloroquine were reinstated. In May 2014, skin sarcoid papules and nodules of the trunk, lupus pernio and sarcoid plaques of the scalp appeared. A biopsy of an abdominal lesion confirmed noncaseating epitheloid granulomas; therefore, methotrexate dosage was increased to 15 mg per week. Again, liver blood tests revealed abnormalities with transaminase, increasing to four times the upper limit of the normal range. The patient refused to resume prednisone treatment and followed only hydrochloroquine treatment at 400 mg per day. In October 2014, a complaint of increased dyspnoea and lupus pernio was noted. Chest radiography (figure 3b) showed bilateral hilar and mediastinal lymphadenopathy, and bilateral infiltrates, with a loss of volume of the right lung. A significant reduction of FVC was noticed on pulmonary function testing (figure 4). Thus, the patient received three methylprednisolone pulses (500 mg each) in December 2014. A month later, in January 2015, there was no clinical or radiological improvement, and FVC was still severely impaired at 1.82 L (47% pred). Her serum ACE level was four times the upper limit of the normal range. Infliximab treatment was started: the patient received 5 mg·kg−1 at week 0, week 2 and every 4 weeks thereafter. After 4 weeks, lupus pernio, scalp and trunk lesions started to improve. These skin improvements preceded a progressive reduction of dyspnoea, improvement of pulmonary function (figure 4), and disappearance of pulmonary bilateral infiltrates and lymphadenopathy on chest radiography (figure 3c). Thoracic HRCT confirmed the presence of signs of fibrosis, with bronchiectasis in the upper lobes and fissural distortion that were not previously present in the 2012 HRCT.
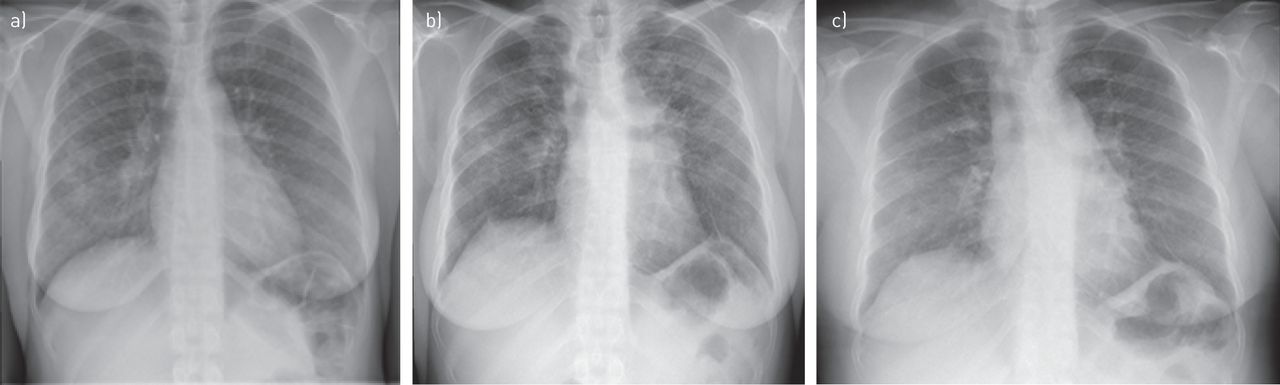
Chest radiography: a) association of pulmonary infiltration and bilateral hilar lymphadenopathy; b) increased infiltration; c) improvement (case 4).
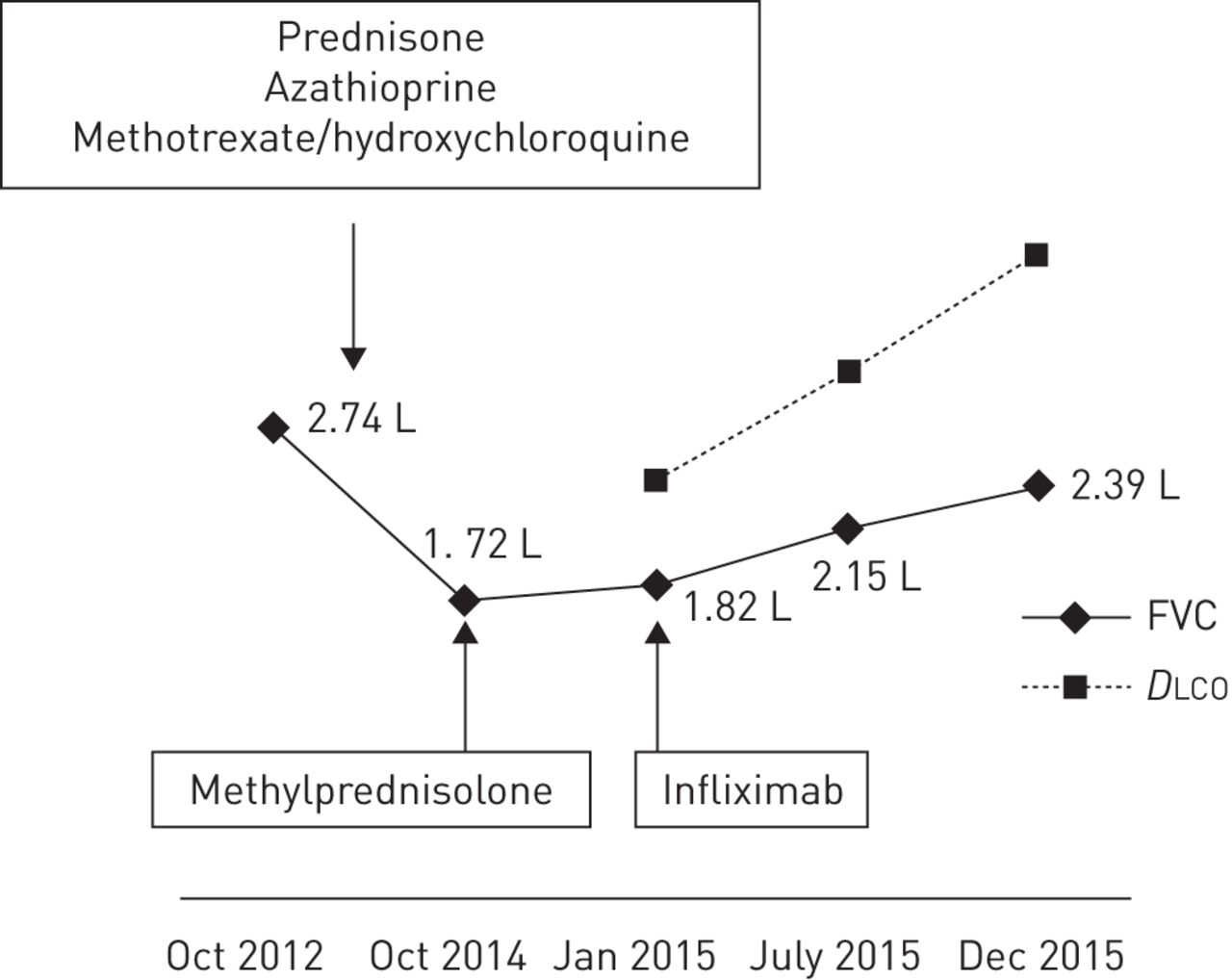
Evolution of pulmonary function tests (case 4). FVC: forced vital capacity; DLCO: diffusing capacity of the lung for carbon monoxide.
Case 5
In 1992, sarcoidosis was diagnosed in a 35-year-old Caucasian man, according to recommended criteria. There was pulmonary fibrosis associated with active lesions, uveitis and hepatic cholestasis. He was treated by steroids but due to having osteoporosis and a prednisone threshold dose higher than 10 mg·day−1, the patient received a combination of steroids with methotrexate, with later azathioprine as a corticosteroid-sparing agent. The absence of efficacy and an intolerance to both immunosuppressive drugs led to their withdrawal. The disease was then controlled by prednisone alone with stable pulmonary function (FVC >90% pred; and FEV1 and DLCO >70% pred, respectively). In October 2003, suspicion of exacerbation of pulmonary sarcoidosis while the treatment was stable led to an investigation. Clinical manifestations included productive cough and increased dyspnoea at exercise. There were no other general symptoms observed, physical examination was normal and thoracic HRCT showed no modification. Pulmonary function testing revealed significant worsening (>10% pred loss of FVC and FEV1) (figure 5). Multiple acid-fast bacilli were found by bronchial fibroscopy and several sputum analyses were made for three consecutive days. Bacterial cultures revealed a Mycobacterium kansasii infection. An antimycobacterial treatment directed against M. kansasii was given, producing significant clinical and functional improvements, with total recovery (figure 5). The outcome was marked by a stability of the lung condition and the occurrence in 2014 of a cardiac localisation with a type 1 atrial–ventricular block (PR interval 270 ms) associated with a significant septal uptake of [18F]fluorodeoxyglucose positron emission tomography (FDG-PET) using a cardiac protocol, with a further control of both cardiac abnormalities on ECG and FDG-PET, thanks to an increasing prednisone dose to 30 mg·day−1. On the last visit in 2015, the patient was in good condition with the sarcoidosis under control, and a continued prednisone dosed of 10 mg·day−1. Thus, this case illustrates a longstanding case of sarcoidosis for which a suspicion of exacerbation of pulmonary involvement led to the discovery of a nontuberculous mycobacterial infection and its subsequent treatment.

Evolution of pulmonary function tests (case 5). Arrow: diagnosis of mycobacterial infection and initiation of antimycobacterial treatment. FVC: forced vital capacity; FEV1: forced expiratory volume in 1 s; DLCO: diffusing capacity of the lung for carbon monoxide.
Discussion
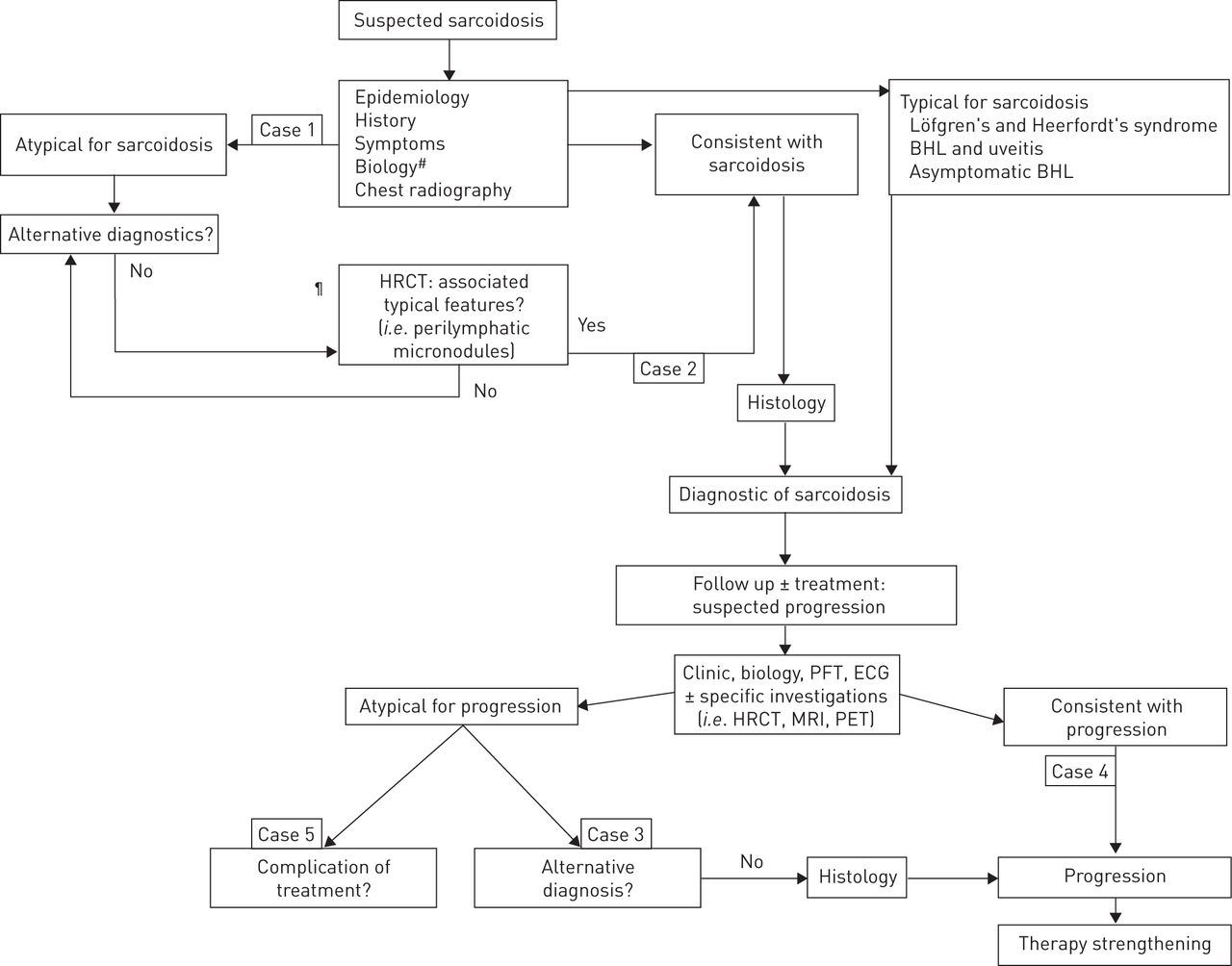
The main clinical problems linked to sarcoidosis are: first, how to reliably confirm the diagnosis; second, how best to identify events occurring during follow-up, particularly new events, and understand whether or not they are specific; and third, confirmation of progression and mechanisms of progression, and eventually how to identify therapeutic response. Each of these points has been well specified in the literature [1–3] but this is not always easy to translate into everyday practice. Thus, the aim of this article was to describe the inherent problems encountered, through emblematic cases, and in so doing, illustrate the best clinical rationale to prevent avoidable mistakes.
First, we focused on positive diagnosis of sarcoidosis at presentation with a discussion of two cases with intrathoracic manifestations: lymphadenopathy and pulmonary infiltration. It is well known that sarcoidosis depends on 1) clinical and radiological presentation, 2) evidence of noncaseating granuloma, and 3) no evidence of an alternative disease. Thus, the first condition to diagnose sarcoidosis is to meet a typical or consistent clinical and radiological presentation. While some typical manifestations, such as Löfgren's syndrome [6], Heerfordt's syndrome, and bilateral hilar lymphadenopathy in a patient with uveitis or no clinical manifestation [17, 18], are easy to identify. It may be more difficult to specify whether or not other manifestations may be either typical or inconsistent. Case 1 is an example of initial misdiagnosis of sarcoidosis. Here, two elements should have been considered. Most importantly, the radiological presentation was very unusual for sarcoidosis, in spite of a typical histopathology, and in the absence of any clinical symptoms, the diagnosis of sarcoidosis should not have been made. In sarcoidosis, thoracic lymphadenopathy is nearly always usual and, as described by several groups [17, 18], left and right hilar lymphadenopathy are observed in 97% of cases. The absence of bilateral hilar lymphadenopathy, with only mediastinal lymphadenopathy appearing in any thorax imaging, is very unusual for sarcoidosis and therefore needs an appropriate search for alternative diagnoses. There are multiple causes of noncaseating granulomatous diseases presented with mediastinal lymph nodes. Besides tuberculosis and histoplasmosis, which may be present without a caseous component, other hypotheses, like common variable immune deficiency, granulomatous lesions associated with past or concomitant carcinoma and lymphomas, are to be considered [19, 20]. Occasionally, hilar lymphadenopathy may be unilateral at presentation but usually it becomes bilateral in the following months. Initially, no blood protein electrophoresis was measured, but this investigation belongs to the diagnostic work-up [21]. A marked hypogammaglobulinaemia, with serum IgG level at 0.32 g·dL−1, serum IgA level at 0.04 g·dL−1 and serum IgM level at 0.011 g·dL−1, was inconsistent with sarcoidosis diagnosis for which serum γ-globulins are either elevated or normal. This indicated that the diagnosis was common variable immune deficiency-associated granulomatosis. This diagnosis can be made in spite of the absence of any history of iterative infections or autoimmune manifestations. Interestingly, in a recent article, Bouvry et al. [19] showed that in common variable immune deficiency-associated granulomatosis, the radiological presentation is often atypical for sarcoidosis with no bilateral hilar lymphadenopathy in most cases despite frequent intrathoracic lymphadenopathy. This case illustrates two lessons: first, it is very critical to analyse particularly the characteristics of thoracic lymphadenopathies, since they are typical in most patients with confirmed sarcoidosis [10], while being present in 70% of patients overall. Second, serum protein electrophoresis is mandatory in the work-up of sarcoidosis at presentation [21]. Case 2 illustrates how to analyse radiological pulmonary infiltration from the perspective of sarcoidosis diagnosis. In this case, diffuse ground glass was predominant. In this context, various alternative diagnoses would have been more possible than sarcoidosis, including hypersensitivity pneumonitis, drug-induced lung diseases, connective tissue-associated lung diseases, desquamative interstitial lung diseases or even Pneumocystis jiroveci infection in immunocompromised patients. However, it has been shown that some elements, particularly associated background HRCT signs, could be very useful to help discriminate between differential diagnoses [22]. Thus, the evidence of either lymphatic micronodules, particularly fissural micronodules, lymphadenopathy or bilateral pulmonary masses strongly suggest sarcoidosis during their presentation [22]. Thus, for case 2, since noncaseous granulomas were in evidence and alternative diagnoses have been ruled out, the diagnosis of sarcoidosis can hereby be confirmed.
Occasionally, both diagnosis and therapeutic problems are intricate, and this may occur in follow-up, as shown in cases 3 and 4. In the case 3, there was certainty with regards to the positive diagnosis of sarcoidosis. However, the occurrence of a significant hypercalcaemia during follow-up, despite not being an uncommon complication of sarcoidosis, appeared paradoxical, subsequent treatment was with significant doses of steroids (30 mg·day−1) in the absence of calcium and vitamin D supplementation. Moreover, the patient had no hypercalcaemia prior the commencement of steroid therapy. In the case of sarcoidosis, hypercalcaemia can be encountered in 5–10% of patients, particularly for Caucasian males over 40 years of age, during summer, on calcium or vitamin D supplementation, or in association with renal sarcoidosis [23–25]. Sarcoidosis is a well-known cause of hypercalcaemia through calcitriol hypersecretion but none of the conditions often associated with hypercalcaemia due to sarcoidosis or increased calcitriol level were evidenced [23]. Moreover, prednisone efficiently prevents intestinal calcium absorption when hypercalcaemia results from sarcoidosis. Thus, in this case, an associated cause of hypercalcaemia, for example, primary hyperparathyroidism, is to be investigated first [26]. In our case, another cause, such as Graves' disease, was eventually shown. Thyrotoxicosis is a well-known cause of hypercalcaemia [27, 28]. Thus, the correct treatment, i.e. treatment of Graves' disease and not a strengthening of sarcoidosis treatment, was indicated and made the normalisation of serum calcium level possible. Case 4 illustrates several points concerning the treatment of patients with sarcoidosis: treatment decision, treatment adverse events, treatment response or resistance. Initially, a treatment with prednisone, the usual first-line treatment in the absence of any contraindication, was initiated according to recommendations, in the context of NYHA functional class II dyspnoea and FVC loss in a biologically active sarcoidosis. Rapid, significant weight gain prompted a decrease in steroid therapy and initiation of azathioprine, which was not sufficient alone. While methotrexate at 20 mg·week−1 in combination with hydroxychloroquine was efficient, it was also responsible for marked cytolysis with withdrawal of methotrexate. Eventually, with a clear progression of dyspnoea, deterioration of pulmonary function and skin flare, third-line therapy with infliximab was given with a slow but marked response at the skin and pulmonary levels. Caution is particularly required with regards to not only the therapeutic response but also adverse events. This case was particularly difficult, with insufficient response to several drugs, serious adverse events with at least two different drugs and eventually a recourse to anti-tumour necrosis factor-α therapy. There was a significant progression of the disease according to symptoms, radiography and pulmonary function. According to Judson et al. [15] and Zappala et al. [16], occasionally, evidencing progression of pulmonary involvement may be difficult. There may not always be a concordant progression according to imaging and pulmonary function. The interobserver agreement for evaluating the disease trend according to chest radiography is far from ideal [29]. It has been shown that pulmonary function, and FVC in particular, is best correlated with the outcome, as shown by the study by Zappala et al. [15].
In case 5, the positive diagnosis of sarcoidosis was confirmed when the disease was at an advanced stage with pulmonary fibrosis, which is seen in 5% of patients at presentation [30, 31]. The disease was still as active as observed in a number of cases with sarcoid pulmonary fibrosis [30]. While the disease had been stable under treatment, certain clues raised the possibility of a sarcoidosis flare, in addition to a cough, and a significant decrease of FVC and FEV1 >10% pred. However, it has been shown, particularly in stage 4, that in the presence of pulmonary worsening, it is necessary to discuss the underlying mechanism, either an associated condition such as an additional infection versus a true exacerbation of sarcoidosis [32, 33]. In the case presented, two elements called for an investigation: first, the occurrence of the event in a patient with a controlled disease with a long-term stable treatment [5, 34]; second, the notion of underlying stage 4 disease with long-duration corticosteroid/immunosuppressive treatment. A diagnosis of an additional infection by M. kansasii was confirmed and on appropriate antimycobacterial treatment without any modification of the sarcoidosis treatment, symptoms were alleviated and pulmonary function restored. It was crucial to avoid a misdiagnosis, which would have led to an inappropriate increase of sarcoidosis treatment and progression of the infection. This case illustrates the importance of considering all possible mechanisms of deterioration in patients with pulmonary sarcoidosis before incriminating a specific exacerbation, particularly in stage 4 and in patients with bronchiectasis [32, 33].
The limitations of this article are linked to the wide scope of sarcoidosis management, hence the choice to consider very practical situations with the potential of overlooking certain important questions. We made the choice to underline many significant day-to-day questions concerning the management of sarcoidosis (figure 6). We focused on the importance of best qualifying the clinical–radiological presentation: typical versus compatible versus inconsistent. We did not consider the evidence of granulomatous lesions even though it is a crucial point. Granulomas must be noncaseous, without exception. Often, one positive specimen is sufficient for diagnosis. The most appropriate biopsies concern superficial lesions (skin, or conjunctive or peripheral lymph nodes) or bronchoscopic specimens (bronchial mucosa, or pulmonary transbronchial biopsies or endobronchial ultrasound-guided transbronchial needle aspiration of mediastinal or hilar lymphadenopathy) [35–37]. This last procedure allows sparing of ∼90% of mediastinoscopies indicated for confirming sarcoidosis [37]. We developed only certain aspects concerning the need to exclude other causes of granulomatosis. The search of these causes is oriented according to exposures (infections, beryllium, etc.) in addition to the presentation of the disease. Another important point we did not consider was the diagnosis of extrathoracic localisations. An important point is that new manifestation during sarcoidosis can be caused solely by sarcoidosis per se and/or other causes. The World Association for Sarcoidosis and Other Granulomatous Disorders recently published tools to use in visceral localisations [38, 39]. We did not consider persistent disabling symptoms such as fatigue, small-fibre neuropathy, cognitive dysfunction, depression and impaired quality of life, which may be at the forefront for many patients with sarcoidosis [40–45]. We did not consider new biomarkers [46] or FDG-PET, which is used only in certain circumstances [47, 48]. Currently, the main uses for FDG-PET are in 1) helping find a site for biopsy in difficult cases, 2) investigation of cardiac sarcoidosis [48], 3) searching for active pulmonary lesions in advanced fibrotic pulmonary sarcoidosis, and 4) searching for metabolic activity in patients with persistent disabling symptoms and normal biomarkers, i.e. serum ACE or serum interleukin-2 [49–51].
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Algorithm for diagnosis and treatment of reported cases. HRCT: high-resolution computed tomography; BHL: bilateral hilar lymphadenopathy; PFT: pulmonary function test; MRI: magnetic resonance imaging; PET: positron emission tomography. #: blood cell counts, blood creatinine, blood calcium, 24-h urinary calcium, aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase, γ-glutamyltransferase, serum protein electrophoresis and angiotensin-converting enzyme; ¶: indications for HRCT were 1) atypical clinical and/or radiographic findings, 2) normal chest radiography but a clinical suspicion of sarcoidosis, and 3) detection of pulmonary complications.
Acknowledgement
The authors acknowledge Nala Shologu for improving the English of the article.
Footnotes
Conflict of interest: Disclosures can be found alongside the online version of this article at err.ersjournals.com
Provenance: Submitted article, peer reviewed
- Received February 23, 2016.
- Accepted April 7, 2016.
- Copyright ©ERS 2016.
ERR articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










