





Abstract
Asthma is a disorder of the airways involving various inflammatory cells and mediators and characterised by bronchial hyperresponsiveness, chronic inflammation and structural alterations in the airways, also known as remodelling. IgE is an important mediator of allergic reactions and has a central role in allergic asthma pathophysiology, as it is implicated in both the early and late phase allergic response. Moreover, clinical and mechanistic evidence has lately emerged, implicating IgE in the development of airway remodelling. The use of monoclonal antibodies targeting IgE, such as omalizumab, has proven very effective in improving respiratory symptoms and quality of life, while reducing asthma exacerbations, emergency room visits and the use of systemic corticosteroids in allergic severe asthma. These effects are believed to be mainly mediated by omalizumab's inhibitory effect on the initiation and further propagation of the allergic inflammation cascade. However, there is evidence to suggest that anti-IgE treatment remains effective long after it has been discontinued. In part, these findings could be attributed to the possible ameliorating effects of anti-IgE treatment on airway remodelling. In this review, we discuss recent findings supporting the notion that anti-IgE treatment modulates the complex immune responses that manifest clinically as asthma and ameliorates airway remodelling changes often observed in allergic severe asthma phenotypes.
Abstract
Anti-IgE treatment suppresses the inflammatory airway response and markers of remodelling in allergic asthma http://ow.ly/Som4i
Introduction
Asthma is a disorder of the airways involving multiple inflammatory cells and mediators [1]. Clinically, it is characterised by symptoms and bronchial hyperresponsiveness and pathologically by chronic inflammation and structural alterations in the airways, also referred to as remodelling. IgE is an important mediator of allergic reactions, including allergic asthma, and has a central role in asthma-related symptoms, airway inflammation and, possibly, airway remodelling [2, 3].
In patients with allergic asthma, exposure to allergens to which they are sensitised results in the rapid release of pro-inflammatory mediators that cause immediate contraction of airway smooth muscle and increased mucus production and the symptoms of the early allergic/asthmatic reaction, that is, wheezing, shortness of breath, chest tightness and cough. Moreover, these pro-inflammatory mediators and chemokines attract and activate inflammatory cells, which release more pro-inflammatory mediators. This slower reaction that takes hours to develop, known as the late allergic/asthmatic response, is characterised by inflammatory infiltration and bronchoconstriction and leads to tissue remodelling.
Remodelling of the airways is a central feature of asthma, especially severe asthma; it can lead to fixed airway obstruction and refractoriness to treatment [4–6]. Most elements of the airway wall are implicated in the structural changes observed during remodelling. Epithelial shedding, reticular basement membrane (RBM) thickening, goblet cell hyperplasia, smooth muscle hyperplasia and hypertrophy, subepithelial fibrosis and angiogenesis are the most prominent findings usually observed in remodelled airways (fig. 1). Although the exact underlying mechanisms of airway remodelling still elude us, the existence of chronic persistent inflammation, involving longstanding exposure of the airways to a variety of environmental agents, cells and mediators, is generally considered a prerequisite [7–9], and IgE is central to the initiation and persistence of this inflammation [2, 10]. Therapeutic agents with the potential to block the allergic cascade at an early stage, such as omalizumab, may potentially also attenuate airway remodelling, a possibility also supported by evidence suggesting that long-term anti-IgE treatment remains effective long after it has been discontinued [11–13].
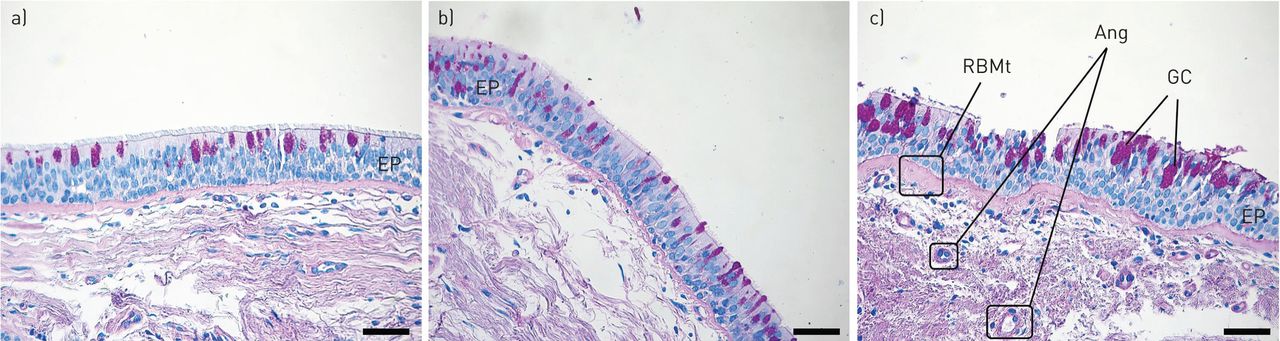
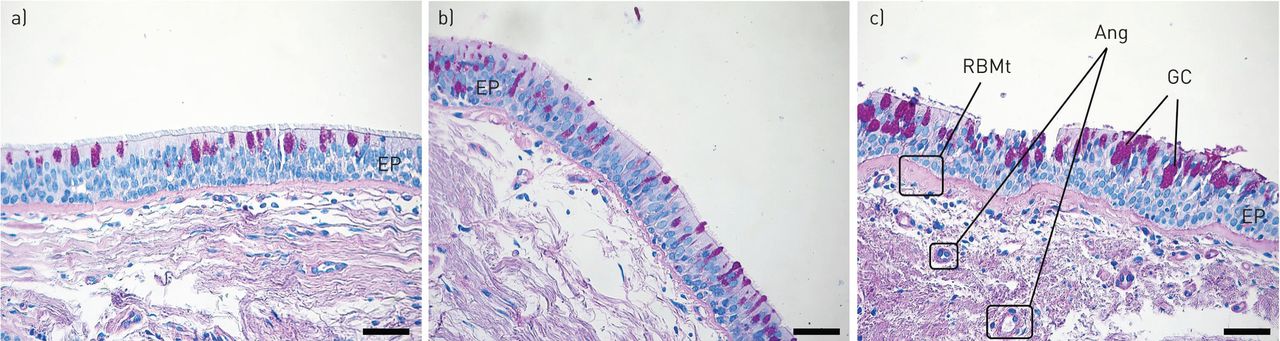{kind=link}
Microphotographs (×20) of endobronchial biopsies from a) healthy control, b) mildly asthmatic and c) severe asthmatic airways demonstrating increased thickness of the basement membrane (haematoxylin–eosin stain) and goblet cell (GC) hyperplasia (periodic acid–Schiff stain) as asthma severity increases. EP: epithelium; RBMt: reticular basement membrane; Ang: angiogenesis. Scale bars=50 μm.
In this review, we discuss recent findings supporting the notion that anti-IgE treatment modulates the complex immune responses that manifest clinically as asthma and reverses airway remodelling changes often observed in severe allergic asthma.
IgE
IgE exists as monomers consisting of two heavy chains (ε-chain) and two light chains, with the ε-chain containing four constant domains (Cε1–Cε4) [14]. Although the main function of IgE is to provide immunity against parasites such as parasitic worms, with the practical elimination of parasites in most of the developed world, IgE is today mainly linked with allergic reactions, playing an essential role in type I hypersensitivity. These reactions include allergic asthma, allergic rhinitis and sinusitis, food allergy and some types of chronic urticaria as well as anaphylactic reactions to certain drugs, allergens and foods.
IgE receptors
There are two kinds of IgE receptors: the low-affinity IgE receptor (FcεRII; CD23) and the high-affinity IgE receptor (FcεRI) [14]. The high-affinity IgE receptors are expressed on the surface of mast cells and basophils as tetramers (αβγ2) and on antigen-presenting cells as trimers (αγ2), at much lower levels [15]. The density of FcεRI expression on human basophils and mast cells correlates directly with IgE levels. Moreover, the expression of the β-chain in FcεRI in mast cells and basophils is upregulated with increased FcεRI expression and amplifies signalling through the receptors. The binding of IgE to the receptor stabilises the receptor on the cell surface [16].
The low-affinity IgE receptor is expressed on B-cells and other haematopoietic cells (T-cells, Langerhans cells, macrophages, monocytes, eosinophils and platelets). The expression of CD23 is upregulated by IgE and interleukin (IL)-4 and CD23 activation mediates IgE regulation, differentiation of B-cells, activation of monocytes and antigen presentation [16, 17].
IgE and the allergic cascade
IgE has a central role in the pathophysiology of allergic inflammation and asthma. Upon first allergen exposure, antigen-presenting cells sensitise naïve T-cells and direct their development towards the T-helper (Th)2 phenotype. IL-4 and IL-13 are produced, increasing FcεRII expression and triggering the relevant B-cells to produce allergen-specific IgE [18, 19]. During their progression to IgE-secreting plasma cells, B-lymphocytes also express membrane-bound IgE, which assists in antigen processing and signal transduction. Secreted IgE binds to the FcεRI on mast cells and basophils and sensitises them to the allergen. Subsequent allergen exposure leads to cross-linking of membrane-bound IgE in mast cells and basophils, inducing degranulation and the release of histamine, tryptase, cysteine-leukotrienes and platelet-activating factors. These mediators have an almost immediate oedematous and bronchoconstrictive effect, key symptoms of the early allergic response [16, 20].
The events driven by the preformed mediators of mast cells and basophils during the early response lead to the production and release of cytokines and chemokines such as IL-3, IL-4, IL-5, Il-13, CC chemokine ligand-5 and granulocyte-macrophage colony-stimulating factor, which recruit neutrophils, eosinophils and basophils, T-cells and macrophages to the site of inflammation [16, 20]. This process, also known as the late allergic/asthmatic response, takes hours to develop and promotes mucus hypersecretion, airway inflammation and hyperresponsiveness. The importance of IgE during late-phase reactions and later establishment of chronic allergic airway inflammation has been shown experimentally in mice lacking the FcεRIα chain, which, compared to wild-type mice, show diminished airway inflammation in an asthma model [21].
IgE and airway remodelling
The activation of the allergic cascade by IgE, under constant allergen stimulation, leads to the establishment of chronic allergic inflammation in the airways of asthmatic patients, with IgE being a key element of the vicious circle that maintains it. Cytokines produced during the late phase and subsequent chronic inflammation stage have been directly associated with the induction of airway remodelling, indirectly implicating IgE in the process. Asthmatic airways are dominated by eosinophils, mast cells and Th2 cells that produce various cytokines, such as IL-4, IL-5 and IL-13, which participate in the perpetuation of airway inflammation and the induction of airway remodelling [22]. IL-4 stimulates synthesis and release of IgE from B-cells and is suggested to be a mediator of airway remodelling by increasing synthesis of α-smooth muscle actin and collagen III [23, 24]. IL-13 induces eosinophilic airway inflammation and promotes mucus metaplasia, subepithelial fibrosis and airway remodelling [25, 26]. Eosinophils also produce and express many fibrogenic factors, particularly transforming growth factor (TGF)-β1 [27]. Finally, other cytokines with increased expression in the airways of asthmatics, such as osteopontin, are closely associated with airway remodelling [28, 29]. It is therefore conceivable that IgE may indirectly contribute to airway remodelling by increasing the production and release of the aforementioned inflammatory mediators.
However, preliminary experimental data suggest that IgE might also be directly associated with airway remodelling (table 1) [30–33]. This would be in accordance with previous studies dissociating chronic inflammation from airway structural changes. Increased RBM thickness has been reported as early as 1 year of age in preschool-aged wheezers [34] and even within 8 days in response to inhaled allergens or to cholinergic stimuli in mild asthmatics [35]. Given the inability of the damaged bronchial epithelium to act as a barrier in severe asthmatics, it is possible for both allergens and IgE to penetrate and directly reach resident tissue-forming cells. Moreover, recent observations suggest that antigen-specific IgE can be produced locally in the bronchial mucosa [36, 37]. Notably, the bronchial epithelial cells of asthmatics express the high-affinity receptor for IgE [38]. Human airway smooth muscle (HASM) cells also express both low- and high-affinity IgE receptors, through which IgE can modulate cellular contraction and inflammatory mediator synthesis, such as IL-4, IL-5 and IL-13 [30, 31]. Interestingly, it was recently demonstrated that IgE stimulation of HASM cells isolated from asthmatic patients in vitro increased cell proliferation and extracellular matrix and collagen deposition in a dose-dependent manner [32, 33].
The main findings of important recent clinical and experimental studies directly associating IgE or anti-IgE treatment with features of airway remodelling
Anti-IgE treatment
Anti-IgE treatment can be mediated by any form of biological medication targeting the synthesis or signalling pathway of IgE, either directly or indirectly, and several such agents are currently under investigation. Lumiliximab (IDEC-152) is a primatised monoclonal antibody against FcεRII that has already been shown to block IgE synthesis in human B-cells and reduce serum IgE levels [39, 40]. Ligelizumab (QGE031), a humanised monoclonal IgG1κ antibody with a high avidity for IgE, was also recently evaluated regarding its pharmacokinetics, pharmacodynamics and safety [41]. Monoclonal antibody-12 is another promising anti-human IgE antibody that may prove useful for the extracorporeal depletion of IgE and IgE-bearing cells from peripheral blood [42]. However, omalizumab is the only biological anti-IgE agent currently licensed for use in humans.
Omalizumab is a recombinant DNA-derived humanised IgG1 monoclonal antibody produced by Chinese hamster ovary cells. It was originally constructed as a murine antibody selectively binding to human IgE [43]. Omalizumab was approved by the US (Food and Drug Administration) in 2003 and by the European Union (European Medicines Agency) in 2005 as an add-on treatment for patients aged >12 years with severe persistent allergic asthma who have a positive skin test or in vitro reactivity to a perennial aeroallergen and who have reduced lung function (forced expiratory volume in 1 s (FEV1) <80%) as well as frequent daytime symptoms or night-time awakenings and who have had multiple documented severe asthma exacerbations despite daily high-dose inhaled corticosteroids, plus a long-acting inhaled β2-agonist. In 2009, it also received approval in Europe for treating patients aged 6–12 years with the same indications without the requirement of reduced lung function.
Omalizumab: how does it work?
The obvious target of any monoclonal antibody against IgE would be circulating (free) IgE. The challenge in creating such an antibody is to avoid cross-linking with cell membrane FcεRI-bound IgE, which would inevitably lead to mast cell and basophil activation and subsequent release of their preformed pro-inflammatory mediators. The design of omalizumab allows it to inhibit the binding of free IgE to the FcεRI by attaching itself to the same antigenic epitope on IgE [44–46]. Conversely, when IgE is bound to the FcεRI on the cell membrane, this epitope is sterically hindered by the receptor and not accessible to omalizumab, therefore blocking its binding and averting any undesirable effects.
Another mechanism by which omalizumab exerts its functions, initially unforeseen by its developers, is its ability to gradually downregulate FcεRI expression on basophils and mast cells [47–49]. This effect is of great importance, as it renders these cells less sensitive to allergen stimulation, leading to a reduction of eosinophilic influx and activation. This negative effect on FcεRI expression was also demonstrated on dendritic cells [50], directly affecting their ability to skew naïve T-cells towards the Th2 pathway and reducing Th2 cytokine release and subsequent inflammation [51]. Interestingly, omalizumab was recently shown to target membrane IgE-bearing B-cells by directly inducing a state of anergy, in which B-cells become unresponsive to antigenic stimuli [52].
All the aforementioned effects of omalizumab have been well documented at a clinical level. In a study of 15 patients allergic to house dust mite, treatment with anti-IgE decreased serum free IgE levels by 99% and FcεRI density on basophils by 97%. Furthermore, a 100-fold increase in antigen dose was needed in order to produce a skin prick test response equal to pretreatment levels [48]. In another study including 24 patients with allergen-induced rhinitis, free IgE levels decreased by 96% and FcεRI expression on basophils decreased by 75% after omalizumab treatment [53]. Reduction in surface IgE and FcεRI expression was also demonstrated in subjects with cat allergy [54]. Moreover, in a recent study of 15 asthmatics who received omalizumab for 12 weeks, basophil FcεRI expression was decreased by 96% while at the same time IL-4, IL-8 and IL-13 FcεRI-mediated production decreased [55].
Clinical data are also available regarding the effects of omalizumab on dendritic cells. Treating patients allergic to cats with omalizumab led to a reduction of FcεRI on blood plasmacytoid dendritic cells and myeloid dendritic cells by 66% and 48%, respectively, while IgE expression was also reduced by >95% [51]. Meanwhile, a modest reduction was also observed in dendritic cell dependent T-cell proliferation to cat allergen after treatment with omalizumab [51]. In another study involving patients with severe allergic asthma, treatment with omalizumab for 16 weeks reduced FcεRI expression on basophils and plasmacytoid dendritic cells by 82% and 44%, respectively [56].
So omalizumab not only reduces circulating free IgE levels but it reduces the expression of its receptors on both effector and antigen-presenting cells, lowering or blocking allergic reactions. In addition, there are several reports suggesting a role for IgE in nonatopic asthma, and some proof-of-concept studies and case reports have also suggested an efficacy of anti-IgE therapy in so-called intrinsic asthma, indicating that anti-IgE treatment may also modulate innate immunity [57].
Omalizumab and airway inflammation
Several clinical controlled studies, as well as real-life experience studies, have demonstrated the safety and efficacy of omalizumab in reducing asthma-related symptoms, corticosteroid use, exacerbation rates and emergency visits, while improving patients' quality of life in inadequately controlled patients with severe allergic asthma [58–61].
These beneficial clinical effects of omalizumab are a result of its profound anti-inflammatory impact on the airways of patients with severe allergic asthma, mediated by the mechanisms mentioned earlier. Numerous studies have shown the effect of omalizumab in the airways of allergic asthmatics. In a hallmark study by Djukanović et al. [62], inflammatory cells were evaluated in induced sputum and bronchial biopsies in 45 steroid-naïve mild/moderate allergic asthmatic patients after 16 weeks of treatment with omalizumab or placebo. Treatment with omalizumab resulted in reduction of sputum and airway eosinophilia. Moreover, omalizumab was associated with a marked decrease in epithelial and subepithelial cells expressing FcεRI and cell-surface IL-4, as well as submucosal B-cells, CD3+, CD4+ and CD8+ T-lymphocytes [62]. In a randomised double-blind placebo-controlled study involving 25 allergic asthmatics, van Rensen et al. [63] provided additional evidence that sputum and airway eosinophils, as well as FcεRI+ and CD4+ cells, are reduced after omalizumab treatment. A few more recent studies reported similar findings [64–66], possibly associated with decreased expression of IL-4 and IL-5 [62, 67].
Omalizumab: a possible modulator of airway remodelling?
By inhibiting IgE-mediated responses, omalizumab disrupts a key element of the allergic inflammatory cascade. Given the crucial role of allergic airway inflammation in the occurrence of structural airway changes, it is conceivable that omalizumab may indirectly contribute to decreased airway remodelling in patients with allergic asthma by inhibiting the expression and production of pro-inflammatory cytokines associated with remodelling, such as TGF-β and endothelin-1 [68, 69]. Limited evidence is currently available to support this notion [3]; however, a few preliminary experimental and clinical studies in the past few years have implicated anti-IgE treatment with the amelioration of airway remodelling parameters, such as airway wall thickening and smooth muscle hyperplasia (table 1).
The first set of data came from an experimental study using a murine model of chronic allergic airway inflammation based on ovalbumin sensitisation and subsequent periodic intranasal re-exposure for 3 months [70]. The authors showed that omalizumab decreased airway hyperresponsiveness and inflammatory cell counts, IL-5 and IL-13 levels in bronchoalveolar lavage fluid. Importantly, omalizumab also decreased markers of remodelling, such as peribronchial collagen III/V deposition, hydroxyproline and α-smooth muscle actin.
Driven by the previous findings on murine models, Roth and colleagues [31, 32] engaged in a series of in vitro experiments utilising primary HASM cells. Initially, they isolated and cultured HASM cells bearing IgE receptors from healthy controls and patients with chronic obstructive pulmonary disease and allergic asthma (n=6 each) [31]. IgE stimulation was reported to increase IL-6, IL-8 and tumour necrosis factor-α mRNA synthesis and corresponding protein secretion in all groups, while omalizumab inhibited cytokine secretion in a dose-dependent manner [31]. The authors continued experimenting with HASM cells isolated from asthmatics and control subjects, demonstrating that IgE induced greater HASM cell proliferation as well as extracellular matrix (ECM), collagen type I, III, VII and fibronectin deposition in asthmatics. Pre-incubation with omalizumab inhibited all remodelling effects [32]. The researchers also found that IgE stimulates collagen type-I and type-VII deposition through IgE receptor-I and extracellular signal-regulated kinase (ERK)1/2 and mitogen-activated protein kinase (MAPK), but the proliferation and the deposition of collagen type-III and fibronectin involves both IgE receptors as well as ERK1/2 and p38 MAPK [32]. Redhu and Gounni [33] later on showed that IgE-induced proliferation of HASM cells is mediated via the MAPK, Akt and signal transducer and activator of transcription-3 signalling pathways.
Based on the results of the previous in vitro studies, two clinical studies set out to investigate the potential effects of omalizumab on the airway wall using computed tomography imaging in a limited number of patients with allergic asthma [65, 66]. The study by Hoshino and Ohtawa [65] was the first to examine the effects of conventional treatment with (n=14) or without (n=16) omalizumab in 30 severely asthmatic patients. Airway wall area (WA) corrected for body surface area (BSA), percentage WA, wall thickness (T)/√BSA and luminal area/BSA were significantly decreased after 16 weeks’ treatment with omalizumab. In the same study, the researchers found a significant reduction in induced sputum eosinophilia from 6% to 2% (p<0.001) and a significant increase in FEV1 after omalizumab treatment, with both changes significantly correlating with changes in WA% [65]. Similar findings were later reported by Tajiri et al. [71].
The aforementioned radiological findings were recently corroborated by histopathological observations of Riccio et al. [64] on bronchial biopsy samples obtained from 11 patients with severe persistent allergic asthma before and after 1 year of treatment with omalizumab. The authors observed a significant reduction in RBM thickness after treatment with omalizumab, accompanied by a marked but not statistically significant reduction in eosinophil infiltration. No correlation was found between reduction in thickness of RBM and reduction in eosinophilic infiltration, possibly attributed to the limited number of patients. Interestingly, a proteomic analysis of bronchial biopsies was also performed by the same group on eight severe asthmatics before and after treatment with omalizumab for 1 year [72]. The authors divided the patients in the basis of whether they responded to treatment and performed a cluster analysis of baseline proteomes to pinpoint differences between groups on the protein level. It was demonstrated that administration of omalizumab downregulated bronchial smooth muscle proteins [72]. Moreover, among all ECM proteins, galectin-3 was the most reliable and predictive biomarker of airway remodelling modulation by omalizumab.
Conclusion
The introduction of biological agents, such as anti-IgE and anti-IL-5 for the treatment of allergic asthma and other allergic diseases, has opened exciting new fields for clinicians to explore [73, 74]. Anti-IgE treatment, as represented by omalizumab, has proven very effective in improving clinical parameters in patients with noncontrolled severe allergic asthma by directly interrupting the vicious circle of inflammation mediated by IgE hypersensitivity. Furthermore, preliminary data support the possibility that anti-IgE treatment may also be a disease modifying pharmacological intervention by directly or indirectly preventing or reversing airway remodelling changes previously considered to be rather permanent. The ongoing EXCELS study (Evaluating Clinical Effectiveness and Long-term Safety in Patients with Moderate-to-Severe Asthma) will shed more information on the natural history of severe asthma and the efficacy of omalizumab [75]. Moreover, there is increasing interest in developing new monoclonal antibodies presenting higher avidity for IgE, such as ligelizumab and lumiliximab, and the ability to interact with low-affinity IgE receptors [76]. Further studies are needed to fully elucidate the exact mechanisms through which anti-IgE treatment benefits patients with severe allergic asthma.
Footnotes
Conflict of interest: Disclosures can be found alongside the online version of this article at err.ersjournals.com
Provenance: Submitted article, peer reviewed.
- Received February 16, 2015.
- Accepted March 5, 2015.
- Copyright ©ERS 2015.
ERR articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References









