





Abstract
Idiopathic pulmonary fibrosis (IPF) is an incurable, progressive interstitial lung disease with a prognosis that is worse than that of many cancers. Epidemiological studies have demonstrated a link between IPF and thrombotic vascular events. Coagulation and fibrinolytic systems play central roles in wound healing and repair, processes hypothesised to be abnormal within the IPF lung. Animal models of pulmonary fibrosis have demonstrated an imbalance between thrombosis and fibrinolysis within the alveolar compartment, a finding that is also observed in IPF patients. A systemic prothrombotic state also occurs in IPF and is associated with increased mortality, but trials of anticoagulation in IPF have provided conflicting results. Differences in methodology, intervention and study populations may contribute to the inconsistent trial outcomes. The new oral anticoagulants have properties that may prove advantageous in targeting both thrombotic risk and progression of lung fibrosis.
Abstract
A review of the coagulation system in IPF exploring scientific and clinical studies in this area http://ow.ly/OdXwX
Introduction
Idiopathic pulmonary fibrosis (IPF) is a devastating disease with a 5-year survival that is worse than that of many cancers. Despite extensive research in recent years, the underlying pathophysiology is incompletely understood, and diagnostic and management decisions are made in the context of a multidisciplinary team [1]. Epidemiological studies have revealed a link between IPF and thrombotic vascular diseases including deep vein thrombosis (DVT), pulmonary embolism (PE) and acute coronary syndromes (ACS) [2–4]. Animal models of fibrosis have demonstrated an imbalance between thrombosis and fibrinolysis, which is also seen in the alveolar compartment in IPF patients [5, 6]. Recent studies have shown a systemic pro-thrombotic state in IPF [7], but clinical trials of anticoagulation have provided conflicting results [8, 9]. This review will discuss the role of the coagulation system in IPF, appraise the findings of the two clinical trials of anticoagulation in IPF and explore a role for new anticoagulants.
Epidemiology of IPF and thrombosis
Hubbard et al. [2] investigated the link between IPF and cardiovascular disease in a population-based study using a large general practice database. They included 920 IPF subjects and 3593 matched controls, and studied the prevalence of cardiovascular disease, cerebrovascular disease and DVT. They reported an increased risk of ACS (OR 1.53, 95% CI 1.15–2.03) and DVT (OR 1.98, 95% CI 1.13–3.48) in subjects who subsequently developed IPF [2]. The risk increased further following IPF diagnosis (rate ratio for ACS: 3.14, 95% CI 2.02–4.87; rate ratio for DVT: 3.39, 95%CI 1.57–7.28).
Studies in Denmark [3] and the USA [4] have confirmed these findings. Sode et al. [3] used national registries to identify 19 557 patients with idiopathic interstitial pneumonia (IIP) and over 7 million controls. The same registries were used to identify subjects with venous thromboembolism (VTE) events. The hazard ratio (HR) for developing IIP in those with VTE was 1.8 (95% CI 1.7–1.9) and was higher in those with PE (HR 2.4, 95% CI 2.3–2.6) than in those with DVT (HR 1.3, 95% CI 1.2–1.4). Again, it was noted that VTE appeared to precede the diagnosis of IIP. To assess the specificity of these observations, Sprunger et al. [4] used US cause-of-death registries to identify IPF cases and three comparison groups: population-based controls, chronic obstructive pulmonary disease (COPD) patients without IPF and lung cancer patients without IPF. A total of 218 991 cases of IPF were identified with a VTE prevalence of 1.74%. This was significantly greater than among population-based controls, for whom the incidence was 1.31% (adjusted OR 1.34, 95% CI 1.29–1.38). A similar increased risk of VTE was associated with IPF compared with COPD (adjusted OR 1.41, 95% CI 1.31–1.46) and lung cancer patients (adjusted OR 1.66, 95% CI 1.61–1.72). VTE was less prevalent in the COPD and lung cancer groups than in the general population, contrasting with previous reports of increased VTE risk in these populations [10].
Despite the inherent limitations of large epidemiological studies, the reproducible demonstration of increased risk of vascular events in IPF patients across three large datasets from different populations provides compelling evidence of an association.
Coagulation and the alveolar compartment
Maintaining vascular integrity and patency is essential to survival, and is achieved through haemostasis. A shift in the balance between thrombus formation and fibrinolysis can lead to pathological vessel occlusion or bleeding. A simplified scheme of the coagulation cascade is presented in figure 1.

A simplified representation of the coagulation cascade. Activation through intrinsic or extrinsic pathways leads to production of thrombin from prothrombin and subsequent conversion of fibrinogen to fibrin monomers that link together to make the stable fibrin clot. The fibrinolytic system breaks down cross-linked fibrin polymers through activation of plasminogen to plasmin. Fibrinolysis is controlled by the balance between plasminogen activators (urokinase and tissue plasminogen activator (tPA)) and plasminogen activator inhibitors (PAI-1 and PAI-2). TF: tissue factor; TFPI: tissue factor pathway inhibitor.
The pro-fibrotic potential of coagulation factors and the role of protease-activated receptors (PARs) in mediating thrombin-induced fibroblast proliferation and extracellular matrix deposition in experimental models of pulmonary fibrosis have been reviewed previously [11].
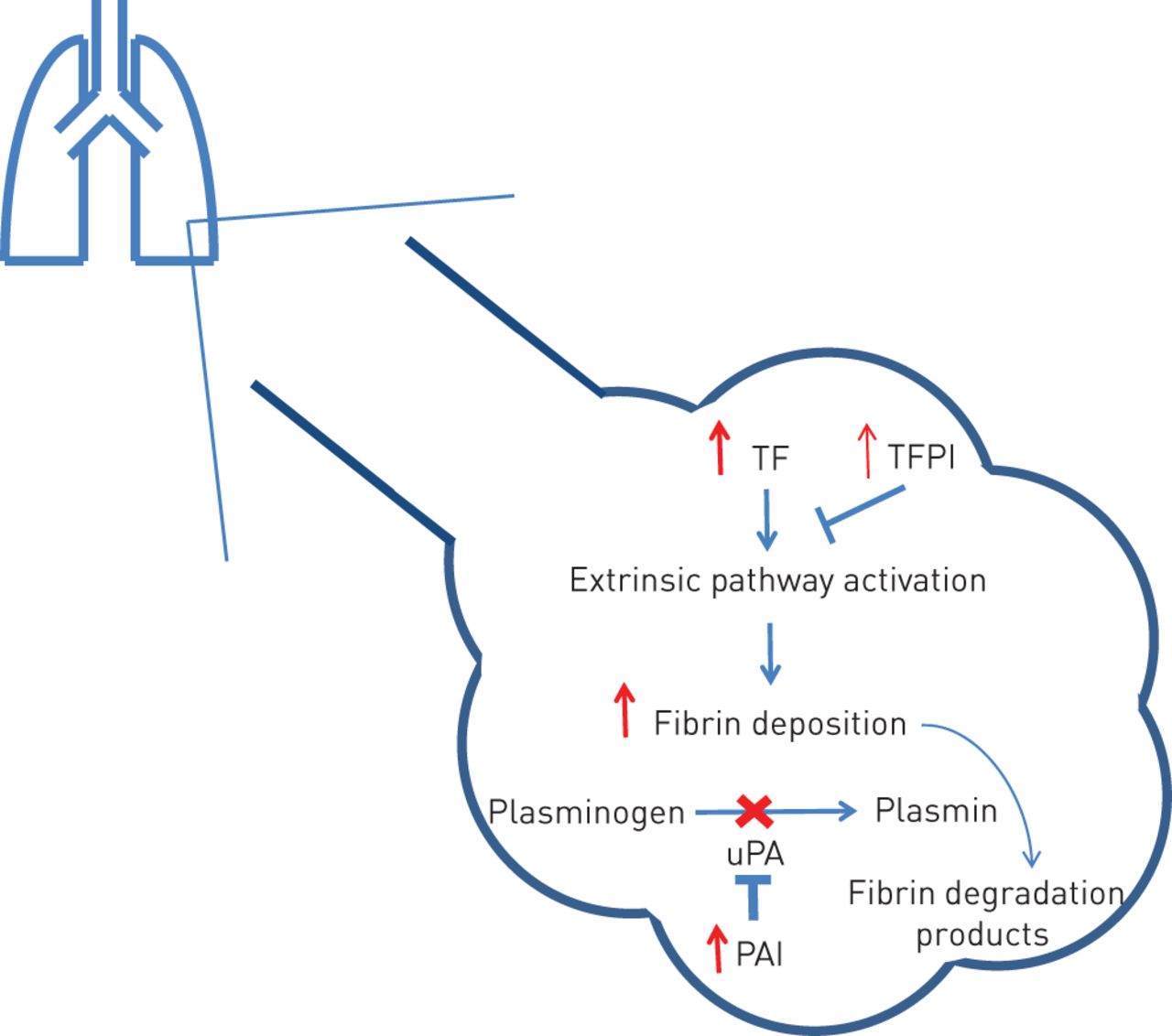
Elevated levels of tissue factor (TF) and plasminogen activator inhibitor (PAI) have been demonstrated in the mouse lung following bleomycin exposure, with alveolar macrophages and type II alveolar epithelial cells implicated in their production [12]. These factors have been studied in the IPF alveolar compartment (fig. 2) [5, 13, 14].

{kind=link}
{kind=link}
The alveolar compartment in idiopathic pulmonary fibrosis (IPF). Tissue factor (TF) is elevated to a greater extent than tissue factor pathway inhibitor (TFPI) resulting in a procoagulant state. In health, urokinase plasminogen activator (uPA) is abundant. However, elevated plasminogen activator inhibitor (PAI) in IPF prevents plasmin production reducing fibrinolysis and favouring fibrin deposition.
TF is a 43-kDa transmembrane cell surface glycoprotein that forms a complex with factor VIIa. Tissue factor pathway inhibitor (TFPI) inhibits the action of TF/factor VIIa complex [5]. TF has been shown to be elevated in bronchoalveolar lavage (BAL) fluid of patients with biopsy proven IPF, with the highest levels seen in patients with severe, progressive disease [5, 6]. Although TFPI is also elevated in the BAL fluid of IPF patients, the balance is in favour of a procoagulant state [5]. Immunostaining revealed TF and TFPI in alveolar epithelial cells and macrophages in IPF patients, but not in controls [5].
Plasmin, produced from plasminogen in response to plasminogen activator, is a broad spectrum protease that is important in fibrinolysis but is also capable of degrading other extracellular proteins that leak into the alveolar space during alveolar injury [15]. In health, there is an abundance of urokinase plasminogen activator (uPA) in the alveolar compartment, favouring fibrinolysis [16]. However, BAL fluid from IPF patients shows increased levels of PAIs [6], with immunostaining demonstrating a similar pattern to that of TF [5, 6, 17]. In the bleomycin mouse model of pulmonary fibrosis, the degree of PAI-1 gene expression positively correlated with lung collagen deposition, supporting a relationship between PAI and fibrosis [15]. Therefore, the alveolar compartment in IPF patients favours activation of the coagulation cascade through elevated TF levels and impaired fibrinolysis. Similar findings have been observed in adult respiratory distress syndrome (ARDS) [16, 18].
Systemic hypercoagulability
Platelets
Platelets circulate in a resting state, becoming activated in response to endogenous agonists (e.g. thrombin, collagen and ADP). In health, platelets rapidly activate and aggregate at the site of vascular injury to maintain vessel integrity but this also occurs in thrombotic vascular disease. In addition to their role in haemostasis, platelets are important in mediating immune responses and tissue repair [19].
The relationship between platelets and the lung in health and in acute lung injury has previously been reviewed [19]. It is important to note that platelet formation and removal occurs within the lung [20, 21], and increased platelet activation has been demonstrated in a number of respiratory diseases including ARDS and COPD [19, 22].
Activation of platelets results in degranulation. Platelet α-granules contain platelet-derived growth factor [23] and transforming growth factor-β [24], both of which are implicated in animal models of pulmonary fibrosis and IPF [25–27]. Mice given intravenous bleomycin exhibited platelet sequestration within the lungs that correlated with collagen deposition, supporting a role for platelets in fibrosis [28]. Indeed, Fahim et al. [29] reported increased platelet–monocyte interactions (a sensitive marker of platelet activation) in patients with IPF compared with non-IPF interstitial lung disease (ILD) and non-ILD controls. Platelets are implicated in the pathogenesis of ARDS, with increased surface P-selectin expression on circulating platelets and demonstration of platelet microthrombi in the pulmonary microvasculature [30]. Acute exacerbations of IPF are associated with a histological pattern indistinguishable from that of the diffuse alveolar damage seen in ARDS or acute lung injury. The demonstration of abnormal platelet responses in stable IPF patients raises the question of whether recurrent occult lung injury is resulting in a low-grade fibrotic response in contrast to the fulminant reaction seen in ARDS. Investigation of platelet responses in IPF patients represents an interesting new avenue of research.
The coagulation cascade and fibrinolysis
The end result of coagulation system activation is fibrin clot formation. Each fibrin monomer is composed of a single E-domain and two D-domains. During fibrin clot formation, a covalent bond is formed between adjacent D-domains, forming the D-dimer antigen. During plasmin-mediated fibrinolysis, the D-dimer antigen is exposed and can be detected using commercially available assays.
Several studies have examined D-dimer concentration in IPF patients. A trial of anticoagulation in IPF measured plasma D-dimer on study entry and reported an elevated mean concentration of 1.9–2.1 μg·mL−1 (reference range <0.5 μg·mL−1) [8]. Among patients admitted with an acute exacerbation of IPF during the study, those who died were noted to have significantly higher levels of elevated D-dimer than those who survived (mean±sd 3.3±2.3 μg·mL−1 versus 0.9±0.7 μg·mL−1; p<0.0001) [8]. A subsequent trial of warfarin in IPF reported mean±sd baseline D-dimer concentrations of 0.9±1.69 μg·mL−1 in the warfarin group and 0.5±0.3 μg·mL−1 in the placebo group [9], although the laboratory reference range and method of analysis were not described (published data report 0.5 μg·mL−1 as the upper limit for VTE exclusion). These studies demonstrate that IPF patients have elevated D-dimer levels indicating increased activation of the coagulation and fibrinolytic systems.
Navaratnam et al. [7] undertook a population-based study to identify if a prothrombotic state was associated with the development and outcome of IPF. They assessed 211 incident IPF cases and 256 matched controls for the presence of a prothrombotic state, which was defined as the presence of at least one out of 11 measured hereditary or acquired coagulation abnormalities. They found that IPF patients were more likely to have a prothrombotic state than controls (OR 4.78, 95% CI 2.93–7.8). Elevated D-dimer was more common in the IPF group (OR 1.69, 95% CI 1.09–2.62), but the striking difference was elevated factor VIII levels (OR 7.02, 95% CI 4.29–11.5). Both of these measures demonstrated a stronger association with IPF at higher levels. Previous smaller studies have demonstrated elevated factor VIIIc, D-dimer, fibrinogen and homocysteine levels in IPF patients, with the highest levels observed in those with an acute exacerbation [31].
Factor VIII exists in the plasma as a complex of proteins. Elevated factor VIII levels are a recognised risk factor for arterial and venous thrombosis [32–35]; therefore, the observation of elevated levels in IPF provides insight into the link with thrombotic vascular disease. However, the nature of the relationship in the lung remains unclear. Elevated factor VIII levels are associated with endothelial injury [32–34] and could occur secondary to the fibrotic process rather than contributing to IPF pathogenesis. The prothrombotic state does appear to impact outcomes in IPF with Navaratnam et al. [7], reporting an associated three-fold increase in mortality.
The utility of stratifying IPF patients based on markers of systemic hypercoagulability is untested, but the observation that a prothrombotic state impacts IPF outcome suggests a potential role in prognostication that may add to existing clinical risk prediction models [36]. Additionally, identifying IPF patients with a prothrombotic state would allow anticoagulation to be targeted to those most likely to benefit. This therapeutic approach may be desirable in IPF [37]. Conducting large clinical trials of individualised therapies in IPF subgroups would require global collaboration in order to achieve adequate statistical power, supporting the need for a global IPF registry [38].
Anticoagulation in IPF
In 2005, Kubo et al. [8] reported an open-label, randomised trial of prednisolone plus anticoagulant therapy versus prednisolone alone in IPF patients. A striking improvement in survival in the anticoagulation group provided hope that treating hypercoagulability in IPF may improve outcomes. However, a subsequent double-blind, randomised, placebo-controlled trial of anticoagulation with warfarin in IPF demonstrated increased mortality in the anticoagulation group [9], with the excess deaths largely due to progressive lung disease. The key features of the studies are outlined in table 1.
Key features of two randomised trials of anticoagulation in idiopathic pulmonary fibrosis
The study by Kubo et al. [8] was significantly limited by its small sample size, lack of blinding, absence of a true placebo-controlled arm and failure to analyse on an intention-to-treat basis, making it difficult to draw conclusions. By contrast, the negative outcome of the larger, randomised, double-blind, placebo-controlled trial of warfarin therapy allows a firm conclusion to be drawn that warfarin therapy is potentially harmful in IPF where there is no other indication for anticoagulation.
Warfarin has been associated with other organ system dysfunction including renal impairment. Warfarin-related nephropathy is characterised by an increase in serum creatinine following periods where the international normalised ratio exceeds the therapeutic range [39]. A case series of nine patients with acute kidney injury and haematuria following warfarin overdose demonstrated widespread glomerular haemorrhage with obstructive red cell casts on biopsy [40]. A larger study of 15 258 patients initiated on warfarin did not demonstrate any increase in haematuria in those that developed nephropathy, indicating that organ dysfunction can occur without overt bleeding [39]. This poses the question of whether the IPF-related deaths in the warfarin group were due to occult alveolar haemorrhage. Although it seems unlikely this would go undetected, it is unclear whether patients in this study underwent post mortem examination [9].
Warfarin interferes with vitamin K metabolism, preventing the production of active (carboxylated) vitamin K-dependent clotting factors. Vitamin K is also essential for the production of the endogenous anticoagulant protein C. Homozygous protein C deficiency results in fatal thrombotic events in infancy and heterozygotes have increased risk of VTE [41]. Initiation of warfarin therapy can result in protein C deficiency before depletion of vitamin K-dependent clotting factors leading to a transient procoagulant state. Protein C also exhibits cytoprotective properties through alteration of inflammatory and apoptotic gene expression, downregulation of release of inflammatory mediators, reduced cell adhesion molecule expression and maintenance of endothelial barrier function [41]. Interference with these protective pathways could contribute to worse outcomes in IPF.
Although methodologically flawed, the magnitude of the signal observed in the study by Kubo et al. [8] is hard to ignore. A notable feature of this study was the use of intravenous low molecular weight heparin during admission for progressive respiratory failure. Indeed, the mortality rate from an acute exacerbation of IPF in the anticoagulation group was comparatively low, at 18% compared with 71% in the non-anticoagulation group [8], and a combined mortality rate of 63% in patients in both groups was seen in the study by Noth et al. [9]. Although the limitations of the study and the high rate of exacerbations make it difficult to generalise these results to the IPF population as a whole, it raises questions about whether warfarin is the correct anticoagulant to be studying in IPF and what is the best time to anticoagulate patients? Some of the issues discussed in this section have been debated in correspondence following the publication of the study by Noth et al. [9] with all parties agreeing that further research is required to examine the role of other anticoagulants in IPF [42].
New therapeutic targets
Two new groups of oral anticoagulants have recently been developed: direct thrombin inhibitors and direct factor Xa inhibitors. At present, in Europe, their use is recommended for specific indications including VTE prophylaxis following hip or knee replacement, stroke prevention in non-valvular atrial fibrillation and treatment of VTE.
Dabigatran is an oral, direct thrombin inhibitor. Thrombin mediates fibrin production from fibrinogen, activates platelets and induces differentiation of fibroblasts into myofibroblasts [43]. In vitro, dabigatran has been shown to inhibit thrombin-induced fibroblast differentiation into myofibroblasts, and reduce connective tissue growth factor and collagen production by lung fibroblasts [44]. Indeed, direct thrombin inhibition has been shown to attenuate lung collagen deposition in the bleomycin mouse model of pulmonary fibrosis [45, 46].
The direct factor Xa inhibitors, apixaban and rivaroxaban, have efficacy in preventing stroke and other systemic emboli in patients with atrial fibrillation, and in the treatment of VTE [47–49]. Factor Xa facilitates the production of thrombin from prothrombin and is increased in fibrotic lung tissue [50]. It has been shown to promote fibroblast proliferation and differentiation in a thrombin-independent manner through PAR-1 signalling, and direct factor Xa inhibition has been shown to attenuate fibrosis in the bleomycin mouse model [50–52].
Therefore, these novel anticoagulants have potential antifibrotic properties in addition to their anticoagulant effects, and their mechanism of action avoids the proposed negative effects of warfarin. Investigation of their potential value in IPF is warranted.
Conclusions
There is compelling epidemiological evidence of an association between thrombotic vascular events and IPF that is supported by biological demonstration of a local and systemic prothrombotic state, which correlates with disease severity and clinical outcomes. While two clinical trials of anticoagulation in IPF have reported conflicting results, it can be concluded that manipulation of the coagulation system plays a role in IPF pathogenesis. Further research is required to delineate the mechanisms underlying the prothrombotic state in IPF, to assess the utility of phenotyping patients based on thrombotic tendency and to evaluate the new classes of anticoagulants in IPF.
Footnotes
Support statement: M.G. Crooks is funded by the UK National Institute for Health Research. No other specific funding was received.
Conflict of interest: None declared.
Provenance: Submitted article, peer reviewed.
- Received September 3, 2014.
- Accepted October 27, 2014.
- Copyright ©ERS 2015.
ERR articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References









