





Abstract
Idiopathic inflammatory myositis (IIM) is a group of rare connective tissue diseases (CTDs) characterised by muscular and extramuscular signs, in which lung involvement is a challenging issue. Interstitial lung disease (ILD) is the hallmark of pulmonary involvement in IIM, and causes morbidity and mortality, resulting in an estimated excess mortality of 50% in some series. Except for inclusion body myositis, these extrapulmonary disorders are associated with the general and visceral involvement frequently found in other CTDs including fever, Raynaud's phenomenon, arthralgia, nonspecific cutaneous modifications and ILD, for which the prevalence is estimated to be up to 65%. Substantial heterogeneity exists within the spectrum of IIMs, and each condition is associated with various frequencies and subtypes of pulmonary involvement. This heterogeneity is partly related to the presence of various autoantibodies encompassing anti-synthetase, anti-MDA5 and anti-PM/Scl. ILD is present in all subsets of IIM including juvenile myositis, but is more frequent in dermatomyositis and overlap myositis. IIM can also be associated with other presentations of respiratory involvement, namely pulmonary arterial hypertension, pleural disease, infections, drug-induced toxicity, malignancy and respiratory muscle weakness. Here, we critically review the current knowledge about adult and juvenile myositis-associated lung disease with a detailed description of therapeutics for chronic and rapidly progressive ILD.
Abstract
Interstitial lung disease is a leading cause of morbidity in dermatomyositis/polymyositis http://ow.ly/M1wB8
Introduction
Idiopathic inflammatory myositis (IIM) [1, 2] is a group of rare connective tissue diseases (CTD) characterised by muscular and extramuscular signs, with lung involvement being a frequent and challenging issue [3–5]. IIM classically encompasses polymyositis, dermatomyositis and inclusion body myositis according to the first classification by Bohan and Peter [1, 2]. Because of the almost total absence of pulmonary involvement in inclusion body myositis, we will focus on dermatomyositis and polymyositis.
The estimated annual incidence of dermatomyositis/polymyositis ranges from 6 to 10 per 1 000 000 [6]. Cutaneous signs, the hallmark of dermatomyositis, often are the initial presentation of disease and may or may not be associated with muscle manifestations. Erythematous rash over the extensor surface of the fingertips (Gottron's sign), eyelids (heliotrope rash), or upper chest or upper back and shoulders (V- or shawl sign) is considered specific of dermatomyositis. Clinically amyopathic dermatomyositis (CADM), or dermatomyositis sine myositis, is defined by the absence of clinically significant muscle symptoms and normal muscle enzymes for periods of >6 months, and is associated with an acute severe form of interstitial lung disease (ILD) [7]. Dissociation between the magnitude of pulmonary and muscular involvement is possible, and physicians may perform paraclinical tests for myositis including elevated muscular enzymes (creatinine kinase and aldolase), abnormal electromyograms, evocative muscle biopsy and an abnormal signal on muscle magnetic resonance imaging. Patients without overt muscular involvement are diagnosed as hypomyopathic dermatomyositis.
Substantial heterogeneity exists within the spectrum of IIMs, and each condition is associated with various frequencies of pulmonary involvement [8]. With the exception of inclusion body myositis, these extrapulmonary disorders are associated with the general and visceral involvement frequently found in other CTDs including fever, Raynaud's phenomenon, arthralgia, nonspecific cutaneous modifications, and lung involvement for which the prevalence is estimated to be up to 65% [8, 9]. The heterogeneity of the clinical spectrum of IIMs is partly related to the presence of various autoantibodies, known as myositis specific autoantibodies (MSA), that target a subset of aminoacyl-tRNA synthetases (ARSs) as well as components of the signal recognition particle (SRP) and nuclear helicase-ATPase Mi2 [10]. Anti-SRP autoantibodies are almost specific for a particular presentation of pauci-inflammatory necrotising myositis, whereas anti-Mi2 is found exclusively in dermatomyositis; both anti-SRP and anti-Mi2 autoantibodies are associated with a paucity of extramuscular features [8]. Anti-SRP autoantibodies are strongly associated with a subtype of acute immune-mediated necrotising myopathy characterised by proximal weakness secondary to pauci-inflammatory myocyte necrosis. Recently, other autoantibodies specific for dermatomyositis have been reported: anti-NXP2, anti-MDA5, anti-SAE and anti-TIF1γ autoantibodies [11]. Additional autoantibodies, including anti-Ro60/SSa, anti-Ro52/TRIM21, anti-La/SSb, anti-U1RNP, PM/Scl and Ku, are frequent in other CTDs associated with myositis and especially in systemic sclerosis (SSc) [12] and are referred to as myositis-associated autoantibodies (MAA) [10].
ILD is the hallmark of pulmonary involvement in IIM, and causes morbidity and mortality, resulting in estimated excess mortality of 50% in some series [13, 14]. ILD may precede the occurrence of muscular signs in up to 20% of cases [15]. ILD is not uncommon in children and was experienced in 19% of juvenile forms of dermatomyositis (JDM) or polymyositis (JPM) [16]. It is present in all subsets of IIM, but is more frequent in dermatomyositis and overlap myositis. dermatomyositis, polymyositis and CADM can also be associated with other presentations of respiratory involvement, namely pulmonary arterial hypertension (PAH) [5], pleural disease [3], infections [17], drug-induced toxicity [18, 19], malignancy (cancer associated myositis (CAM)) [1, 2] and respiratory muscle weakness.
Here, we critically review the current knowledge about myositis-associated lung disease.
Classification of idiopathic inflammatory myositis
Several diagnostic criteria for IIM have been proposed. This section details four approaches to the classification of IIM.
Bohan and Peter's criteria
The criteria described by Bohan and Peter [1, 2], in 1975, probably remain the most widely used, despite the discovery of numerous autoantibodies and the refinement of histopathological techniques (table 1) [20]. Bohan and Peter's classification encompasses primary polymyositis, primary dermatomyositis, CAM, childhood dermatomyositis or polymyositis, and dermatomyositis or polymyositis associated with other CTDs (overlap myositis) [1]. This classification has severe limitations, which have been highlighted by several authors and include its development in a single centre, the absence of clear instructions to rule out other forms of myopathies (especially inclusion body myopathy), the lack of specific criteria for what should be considered a typical dermatomyositis rash, and the absence of external validation with estimation of sensitivity or specificity [21].
Classifications of idiopathic inflammatory myositis
Classification based on autoantibodies
In 1991, Love et al. [22] proposed a new approach to classifying IIMs based on the description of clinical features associated with MSAs, classically encompassing anti-ARS, anti-SRP and anti-Mi2 autoantibodies (table 2). The anti-ARS autoantibodies identified a subset of patients with frequent ILD and relapsing myositis. The patients with anti-SRP autoantibodies presented with severe myositis, contrasting with those with anti-Mi2 antibodies who had dermatomyositis with a good prognosis [22]. This approach was widely followed by many research groups and four pulmonary phenotypes related to distinct autoantibodies will be described in this review.
Auto-antibodies in dermatomyositis and polymyositis with corresponding clinical spectrum
Anti-tRNA-synthetase autoantibodies-associated ILD
The spectrum of disease associated with anti-ARS autoantibodies was first described in 1990 [23]. Anti-ARS autoantibodies are the most frequent autoantibodies in patients with dermatomyositis (20%) and polymyositis (29%) (table 2) [8]. Anti-ARS autoantibodies are considered specific for IIMs and recognise a subset of ten ARSs [10, 24–28]. Out of all the anti-ARS autoantibodies identified (listed in table 2), anti-Jo1 (anti-histidyl) antibodies are the most commonly found. Anti-PL7 (anti-threonyl) and anti-PL12 (anti-analyl) antibodies are less frequent, whereas the others are exceedingly rare and are not commonly tested in autoimmunity laboratories. Several studies have shown an increased risk of ILD, arthritis, Raynaud's phenomenon, mechanic's hand and fever in patients with myositis and anti-ARS autoantibodies as compared with seronegative controls, an association that is often referred to as the antisynthetase syndrome.
Anti-PM/Scl autoantibodies-associated ILD
Anti-PM/Scl autoantibodies were first identified in 1977 [29] and are directed against a nucleolar macromolecular complex of peptides of 75 kDa (PM/Scl 75 protein) and 100 kDa (PM/Scl 100 protein), with the former being considered the main autoantigen (table 2) [30]. Anti-PM/Scl autoantibodies have been found in 17% of patients with IIM–limited SSc overlap syndrome, compared with 7% of patients with isolated SSc [31], 9% of dermatomyositis patients and 6% of polymyositis patients [8]. Thus, anti-PM/Scl autoantibodies are usually classified as MAA (table 2). Pulmonary symptoms are concomitant of the other CTD manifestations in 11–58% of patients, and occur after the CTD features in 41–55% [19, 32]. The occurrence of ILD before the cutaneous and muscular manifestations of polymyositis or dermatomyositis is less frequently encountered than in subjects with anti-ARS autoantibodies [15, 19, 32].
Anti-MDA5 autoantibodies-related ILD
In 2005, Sato et al. [33] described an autoantibody targeting RNA helicase encoded by melanoma differentiation-associated gene 5 (anti-MDA5) that delineates a distinctive clinical pattern with rapidly progressive ILD and CADM with cutaneous ulcers. The anti-MDA5 autoantibodies are specific for dermatomyositis, and may be found in 7–13% of adult dermatomyositis patients (table 2) [34–36]. Clinically significant myositis is present in only 20% of patients with anti-MDA5 autoantibodies [37]. In a large European cohort of 285 childhood-onset IIM patients, anti-MDA5 autoantibodies were found in 7% of patients (table 2) and were associated with a higher frequency of mild muscular involvement and of radiological evidence of ILD (19%) [38].
Anti-NXP2 autoantibodies and ILD
Anti-NXP2 autoantibodies were first reported in JDM [39], and are less frequently found in adult patients with dermatomyositis, with a wide range of prevalence estimates (1.6–30%) averaging as 17% of dermatomyositis patients (table 2) [40–42]. In a cohort of adult patients with IIM [43], anti-NXP2 autoantibodies were the most prevalent antibodies, followed by anti-Jo1 autoantibodies, and were mutually exclusive of other MSAs. One recent case of ILD with subacute onset of dyspnoea and organising pneumonia has been reported in a patient with dermatomyositis associated with anti-NXP2 autoantibodies, suggesting that these markers should be included in the list of diagnostic tests in patients with ILD and suspected IIM [44].
Classification according to extramuscular features
Two recent studies by Troyanov and co-workers [11, 45] provided significant advances in the classification of IIMs. The first study introduced the concepts of “pure” polymyositis with poor muscular prognosis, and of overlap myositis defined by the occurrence of extramuscular signs with MSA/MAA autoantibodies (table 1) [45]. The frequency of polymyositis was only 9%, while the frequency of overlap myositis was 67% of the series. The second study described two phenotypes of dermatomyositis: pure dermatomyositis (dermatomyositis as the first manifestation, with high cutaneous score, chronic and recurrent cutaneous flares, no mechanic's hands or extramuscular involvement, presence of dermatomyositis-specific autoantibodies, perifascicular atrophy at biopsy and possible paraneoplastic aetiology) and overlap myositis with dermatomyositis features that are associated with poor survival (myositis as the first manifestation, isolated features of dermatomyositis rash, mechanic's hands, and anti-ARS, anti-PM/Scl autoantibodies or MSA) [11].
According to the definition of Troyanov and co-workers [11, 45], all patients presenting with IIM associated with ILD are classified as overlap myositis without distinction between clinical subtypes or prognosis. For this reason, this classification is of little value in the management of IIM-related ILD.
Classification based on muscle histopathology
Finally, the more complex and ambitious classification of the 119th European Neuromuscular Centre (ENMC) workshop (2003) includes inclusion body myositis, definite polymyositis, probable polymyositis, definite dermatomyositis, probable dermatomyositis, CADM, nonspecific myositis, and immune-mediated necrotising myopathy or necrotising autoimmune myopathy related to anti-SRP or 3-hydroxy-3-methylglutaryl-coenzyme A reductase autoantibodies (table 1) [46]. Unlike to the two other classifications based on clinical features and autoantibodies, the ENMC classification system is based on the use of muscle biopsy for classifying patients [46].
Overall, the classification of IIMs has evolved to better reflect the clinical and biological correlations of this spectrum of disease. The patients with IIMs and ILD seen by respiratory physicians mostly fall into the categories of dermatomyositis, classic polymyositis according Bohan and Peter's classification (or overlap myositis as defined by Troyanov and co-workers), and CADM.
Types of pulmonary involvement
A patient with pulmonary signs or complaints may suffer from a variety of chest manifestations in the setting of dermatomyositis/polymyositis. Physicians have to consider the following: direct pulmonary involvement (listed in order of decreasing frequency: ILD, group 1 or group 3 pulmonary hypertension, combined pulmonary fibrosis and emphysema (CPFE) syndrome, and pleural involvement); indirect pulmonary complications (infection, drug-induced lung disease, cancer, respiratory muscle weakness, congestive heart failure, etc.); and the possible combinations or associations with any of the mechanisms mentioned earlier.
Direct pulmonary involvement
Interstitial lung disease
Epidemiology
ILD is a frequent manifestation of dermatomyositis/polymyositis with the prevalence of clinically relevant ILD ranging from 17% to 36% [9, 47–51]. In a prospective series of patients with systematic assessment of lung involvement, the prevalence was estimated to be 65% [9]. ILD occurs after the diagnosis of polymyositis or dermatomyositis in up to 40% of patients, and precedes the diagnosis of overt CTD in 20–30% of cases [47, 52]. In large cohorts of JDM/JPM, ILD occurs in 8–13% of patients and thus seems to occur less frequently than in adult dermatomyositis [53, 54]. Risk factors for ILD in a patient with dermatomyositis/polymyositis include genetic predisposition [55, 56], some clinical signs such as arthralgia [47], and autoimmune features (table 2) [22, 56, 57]. After muscular involvement, dermatomyositis/polymyositis-associated ILD is the second major contributor to morbidity [47]. Up to half of patients with IIM-associated ILD may eventually die from respiratory failure [13]. The mortality rate is especially high in acute forms of ILD despite immediate and aggressive treatment [48, 58].
Clinical presentations
Usual presentation
The signs and symptoms related to ILD in IIM do not differ from idiopathic ILD, and include dyspnoea on exertion, cough, decreased exercise tolerance, digital clubbing and asthenia. Physicians who diagnose ILD have to pay particular attention to the extrapulmonary features of IIMs to distinguish secondary ILD from primary ILD. The assessment of such patients encompasses careful general (weight loss and fever), cutaneous (rash, sclerodactyly, Raynaud's phenomenon, calcinosis, cutaneous ulcers and mechanic's hands), muscular (loss of muscle mass, muscular testing and myalgia), and rheumatologic (arthralgia, arthritis and Jaccoud's deformity) examination. Subcutaneous calcinosis is present in ∼25% of cases of JDM, in contrast to its rarity in adult onset dermatomyositis.
Beyond clinical assessment, the diagnosis of hypomyopathic forms of myositis requires creatine kinase and aldolase testing. In cases of high suspicion of IIM-related ILD (e.g. rash evocative of dermatomyositis or MSA positivity), supplementary investigations adapted for each patient comprise electromyography, muscular resonance magnetic imaging and muscle biopsy.
Testing for the presence of antinuclear autoantibodies completes the clinical evaluation; further testing for MSA or MAA depends on the pattern of nuclear fluorescence in most instances. The clinical spectrum associated with anti-ARS autoantibodies is highly heterogeneous and includes arthritis (62%), mechanic's hand (28%), fever (43%), Raynaud's phenomenon (47%) (fig. 1), myositis (57%) and ILD (70%) in its classical description [8, 15, 23]. Because of the high frequencies of nonspecific signs associated with anti-ARS autoantibodies (i.e. Raynaud's phenomenon, fever, hyperkeratosis and cracking of the skin on the fingers), we favour a revised definition of anti-ARS syndrome that was proposed by Connors et al. [51] based on major (arthritis, ILD and myositis) and minor criteria (table 3). In a meta-analysis of clinical manifestations, the frequency of ILD and fever was increased by 20–30% in patients with anti-ARS autoantibodies other than anti-Jo1 autoantibodies (especially anti-PL7 and anti-PL12 antibodies) as compared with those with anti-Jo1 autoantibodies [8]. By contrast, the frequency of mechanic's hands and of articular and muscular involvement was increased by 50% in patients with anti-Jo1 autoantibodies as compared with other anti-ARS antibodies [8]. Patients with anti-PL7 or anti-PL12 antibodies frequently present with isolated ILD, and are therefore mostly seen by respiratory specialists, whereas other signs are missing [15]. Patients with double positivity for anti-ARS and anti-U1RNP antibodies frequently experience manifestations of dermatomyositis (55%) and Raynaud's phenomenon (82%) [15]. In patients with anti-ARS antibodies, the presence of mechanic's hands may be associated with a higher risk of developing ILD [59].
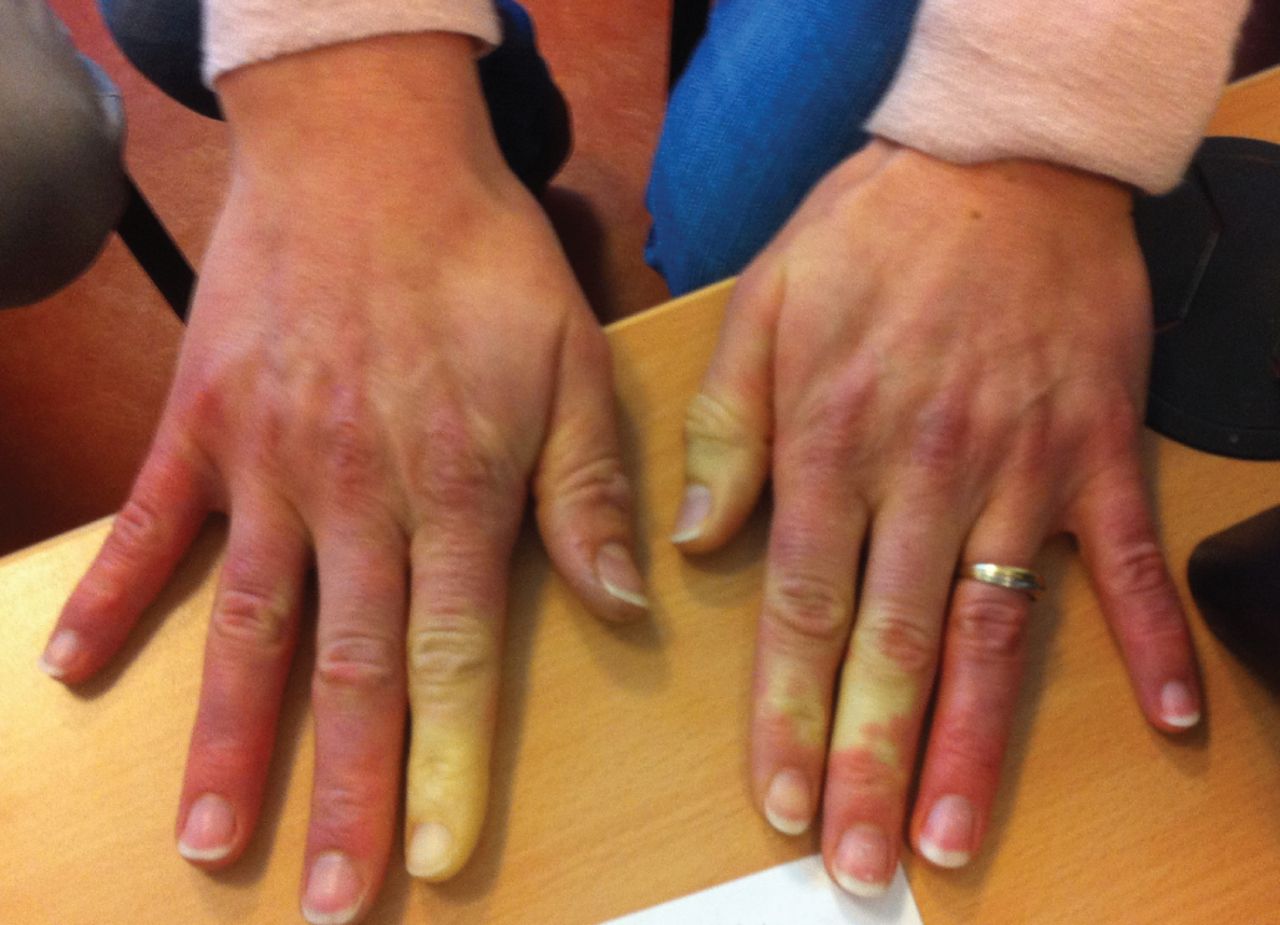
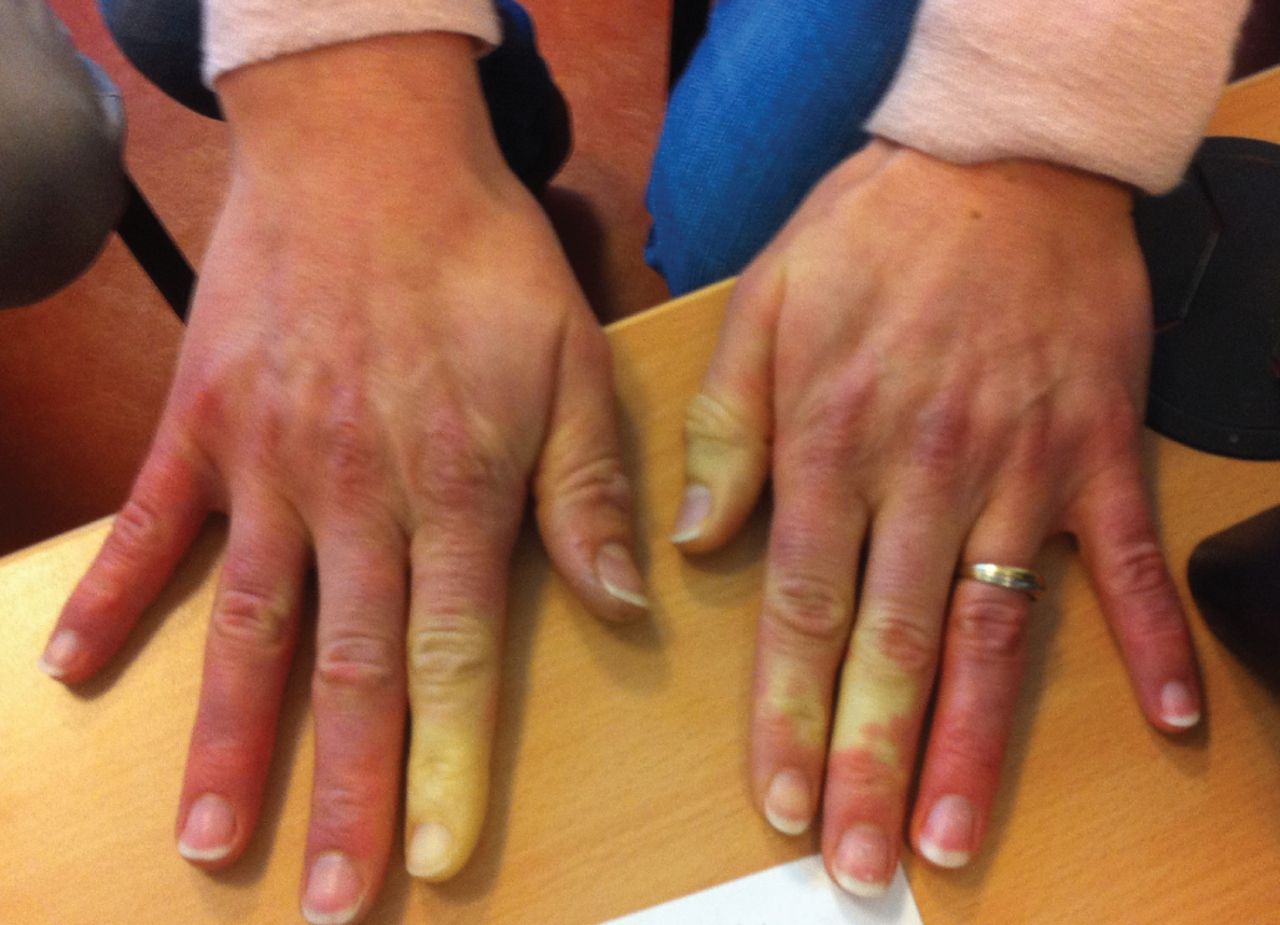
Raynaud's phenomenon in a patient positive for anti-PL7 antibodies.
Proposed criteria for myositis associated with anti-tRNA synthetase antibody
The clinical features described in patients with anti-PM/Scl antibodies largely overlap with those in patients with anti-ARS autoantibodies [19, 32]. Indeed, anti-PM/Scl positive patients commonly experience arthralgia (74%), Raynaud's phenomenon (63%), ILD (38%), fever (33%) and mechanic's hands (11%) [8]. Cutaneous manifestations of dermatomyositis and sclerodactyly are reported in 49% and 76% of cases, respectively [8]. The rash is usually mild and transient, with isolated heliotrope rash or Gottron's papules [11].
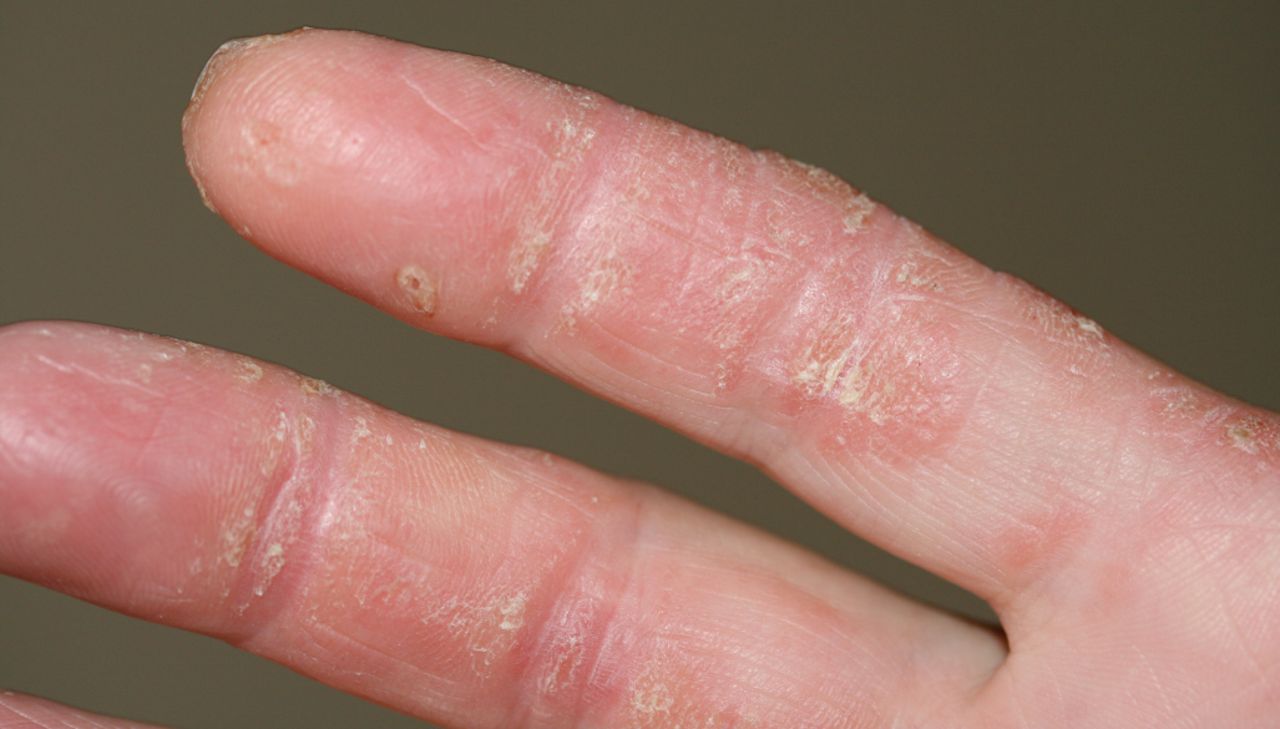
In cases with the presence of anti-MDA5 autoantibodies, digital ulcers are located mainly over the Gottron's papules and digital pulps or periungueal areas, but can occur in other sites of the dermatomyositis rash, i.e. the dorsal side of the elbows, knees, auricles or buttocks (fig. 2) [57, 58]. One European study reported an increased frequency of panniculitis [56]. Arthritis (80%), Raynaud's phenomenon (45%) and mechanic's hands (80%) (fig. 3) are frequently found [55]. Anti-MDA-5 autoantibodies have now been added to the differential diagnoses to consider, especially when the clinical features would suggest anti-ARS or anti-PM/Scl autoantibodies in dermatomyositis patients [55]. Importantly, screening for antinuclear autoantibodies may be negative in patients with anti-MDA5 autoantibodies, which therefore have to be measured specifically upon clinical suspicion [56].

Clinical features associated with anti-MDA5 autoantibodies include severe cutaneous ulcers of a, b) the fingers and c) the dorsal side of the elbow.
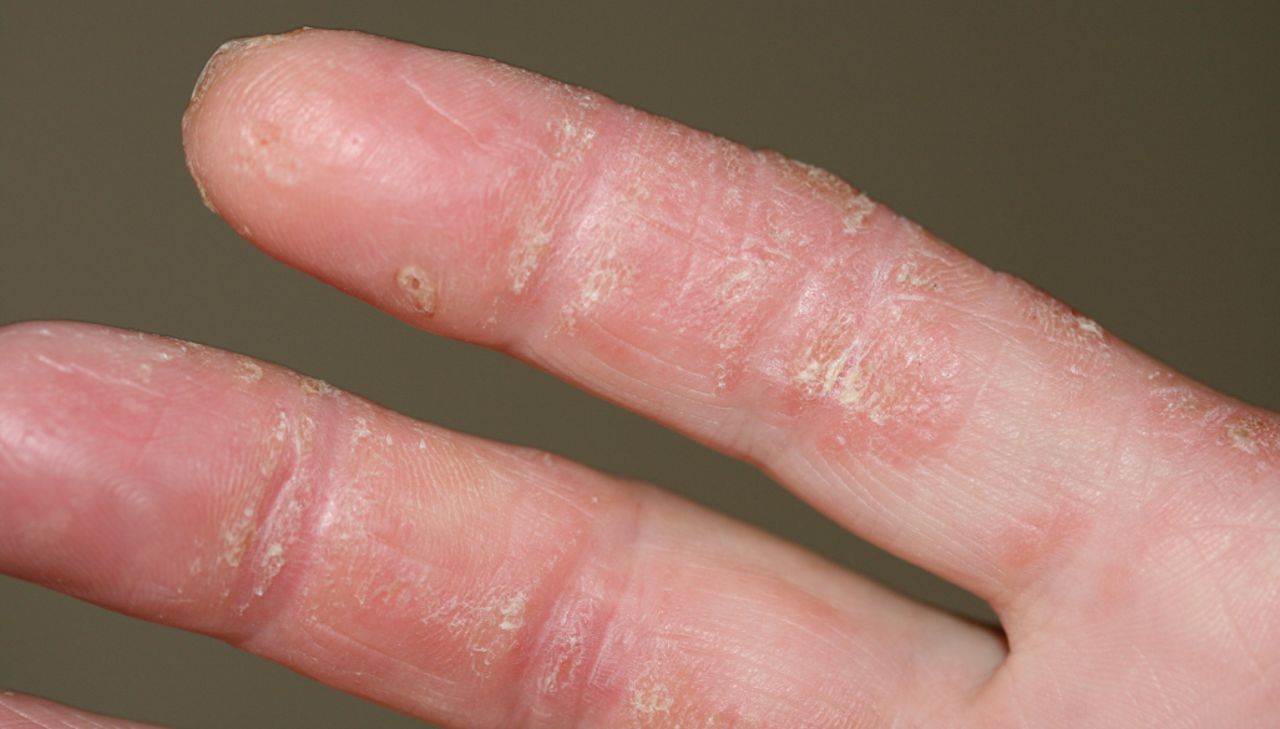
Mechanic's hands in a patient with anti-MAD5 positive dermatomyositis. Image courtesy of Sébastien Debarbieux (Dept of Dermatology, Centre Hospitalier Lyon Sud, Lyon, France).
Rapidly progressive ILD
Rapidly progressive ILD is defined by an acute interstitial pneumonia progressing over several weeks or a few months. Among all CTDs, this aggressive presentation of ILD is suggestive of IIM and especially of dermatomyositis, CADM or hypomyopathic dermatomyositis [60, 61]. Thus, physicians have to search thoroughly for minimal skin modifications or mild muscular involvement in the setting of rapidly progressive ILD.
The presence of anti-ARS autoantibodies seems relatively protective against rapidly progressive ILD, although this presentation has been reported [62, 63]. The detection of anti-MDA5 autoantibodies identifies dermatomyositis patients with a high risk of developing rapidly progressive ILD, with a pooled sensitivity of 77% and a specificity of 86% in a meta-analysis of 631 dermatomyositis or polymyositis patients [64, 65]. Fulminant presentations with ILD, CADM and anti-MDA-5 autoantibodies may be especially frequent in Japanese patients compared with other east Asian or non-Asian patients [35, 64], although a publication bias cannot be excluded. In a Japanese retrospective series of 54 patients with JDM [66], 10 had rapidly progressive ILD and 19 had chronic ILD, illustrating the particularly frequent pulmonary involvement in the Asian population.
Pathogenesis
Aside from recent advances in patients with anti-MDA5 autoantibodies, the pathogenesis of dermatomyositis- or polymyositis-related ILD is poorly known, contrary to that of SSc [67]. Immunohistochemistry on muscle biopsy provides some clues as to immune mediated mechanisms. CD8+ T-cells are usually found in polymyositis with a diffuse cytotoxic effect leading to muscle cell necrosis [68], whereas B-cells and CD4+ T-cells predominate in perivascular areas in dermatomyositis and may be responsible for muscular vasculitis [67, 69]. In lung biopsy specimens of patients suffering from dermatomyositis- or polymyositis-associated ILD, numerous activated CD8+ lymphocytes are found in both affected and relatively preserved tissue. The proportion of these cells in the bronchoalveolar lavage (BAL) fluid might predict the outcome after treatment with corticosteroids, as CD8+ T-cells are significantly increased in corticosteroid-resistant cases of dermatomyositis- or polymyositis-associated ILD [70]. This observation needs confirmation, however, before it can be implemented in the clinic.
In addition, correlations are emerging between some genotypes and the clinical phenotype or prognosis. For example, a polymorphism in the promoter of the tumour necrosis factor-α gene is increased in dermatomyositis/polymyositis patients with ILD (OR 2.1, 95% CI 1.0–4.3) [71]. The HLA-DRB1*03 haplotype, a determinant factor in the development of ILD among patients with CTDs [72, 73], predicts a higher risk of ILD in dermatomyositis/polymyositis [74], irrespective of the myositis subtype or the presence of autoantibodies [56]. Anti-PM/Scl and anti-ARS antibodies also share the same prevalence of HLA-DRB1*03, DQA1*05 and DQB1*02 haplotypes [75, 76].
Recently, gain of function mutations in the IFIH1 gene encoding the protein MDA5 were reported in association with various inflammatory conditions including a skin rash reminiscent of JDM skin lesions [77]. Anti-MDA5 is a cytosolic double strand RNA receptor, and gain of function mutations of IFIH1 lead to enhanced type I interferon production. Similarly, gain of function mutations in the TMEN173 gene encoding the DNA sensor STING (stimulator of interferon genes), which is responsible for enhanced type I interferon production, were found in patients with vascular involvement and severe ILD [78]. Taken together, these findings suggest that type I interferon may be a key cytokine linking anti-MDA5 autoantibodies to lung involvement and ILD.
Radiology
High-resolution computed tomography (HRCT) is essential for evaluation of ILD. Pulmonary HRCT scanning is a noninvasive and sensitive test, demonstrating evidence of pulmonary changes in all patients suffering from ILD. These changes are predominantly localised in the lower lobes, as reported by previous studies [47, 79]. Linear opacities, ground-glass attenuation (hyperattenuated areas in which the bronchi and vessels remain visible), reticulation and peribronchovascular thickening are the most common HRCT abnormalities at the initial imaging of ILD. Consolidation corresponding to areas with increased attenuation and decreased visibility of bronchial walls and vessels are commonly found, and correspond pathologically to areas of organising pneumonia. A combination of patchy areas of consolidation and reticulation is frequent, and suggestive of the diagnosis of myositis in subjects with ILD, especially of subacute or acute onset. Honeycombing (areas of small cystic spaces with thickened walls), traction bronchiectasis (bronchial dilation due to traction by fibrous tissue) and subpleural bands are less frequently observed [80].
Pathologic and imaging correlations in IIM are generally similar to those in the idiopathic setting. Overall, a variety of HRCT patterns may be observed in IIM-associated ILD, with especially those of organising pneumonia, nonspecific interstitial pneumonia (NSIP) (fig. 4) or mixed NSIP–organising pneumonia pattern, whereas the usual interstitial pneumonia (UIP) pattern (fig. 5) is less frequent [19]. However, studies that correlate imaging to pathology in the context of IIM are limited and suffer from selection bias, as clinicians tend to find lung biopsy indicated in the less typical cases. Therefore, caution must be exerted when interpreting the imaging, and HRCT patterns cannot be directly transposed into histological considerations.

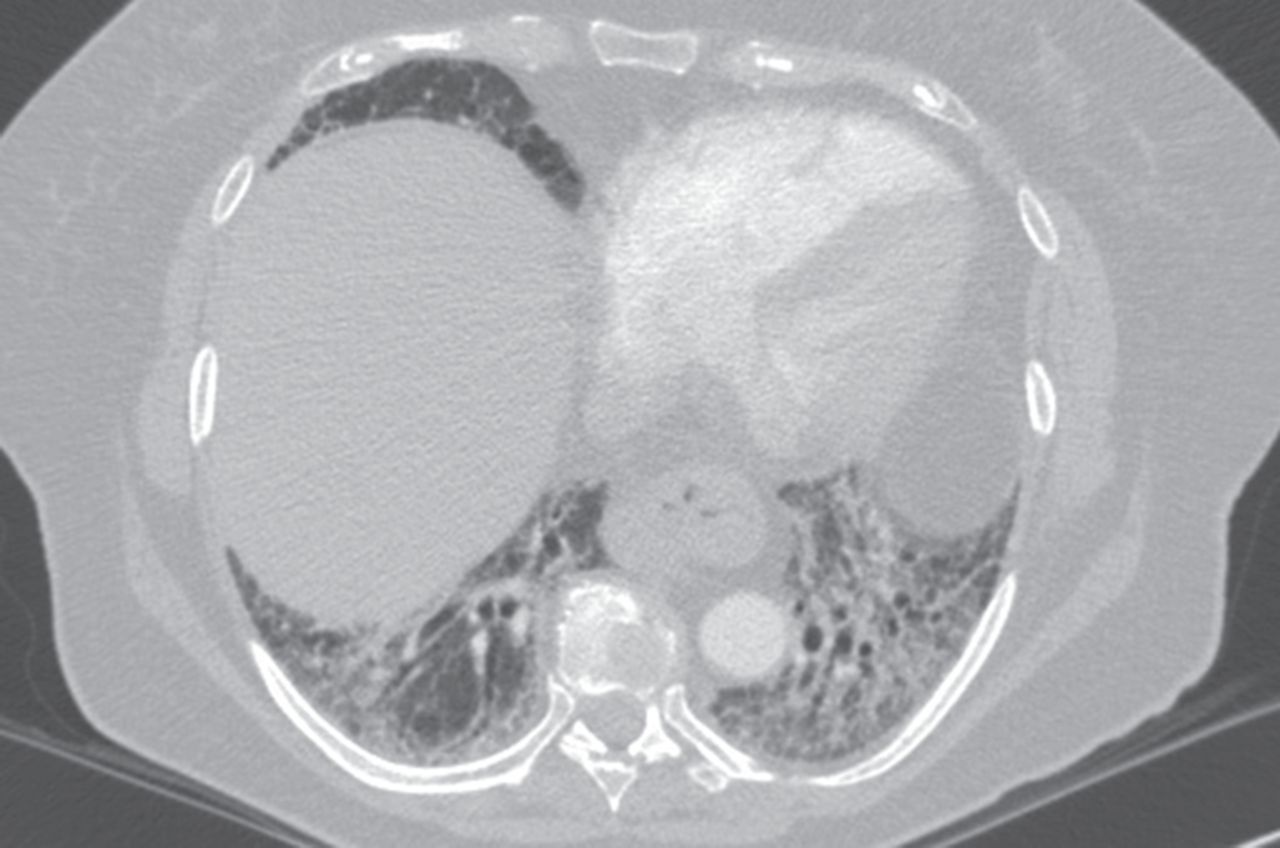
High-resolution computed tomography from a patient with anti-Jo1 positive polymyositis showing basilar predominate reticulation and ground-glass opacity without honeycombing, in a pattern suggestive of nonspecific interstitial pneumonia.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
High-resolution computed tomography from a patient with polymyositis showing honeycombing in a pattern consistent with usual interstitial pneumonia.
HRCT findings do not differ significantly between the various presentations of polymyositis- or dermatomyositis-ILD [81]; however, acute and subacute onset of ILD seem to be associated with more frequent and more extensive consolidation, which is the hallmark of this ILD subtype. Other HRCT features include diffuse patchy ground-glass opacity, basal irregular lines and some reticulation [63]. HRCT patterns at diagnosis can help predict the prognosis of patients with polymyositis- or dermatomyositis-ILD [82], and consolidation (mostly associated with a histological pattern of organising pneumonia) is generally reversible with treatment.
Polymyositis-ILD may be associated with a better prognosis than other forms. The HRCT patterns of dermatomyositis-ILD can be categorised in three groups to help clinical decisions [83]. Dominant ground-glass attenuation and/or reticular opacity without honeycombing, subpleural bands, and traction bronchectasis are often associated with severe ILD and a possibly fatal outcome. Prominent consolidation is typical of acute onset ILD that responds well to corticosteroids and often immunosuppressive therapy [84]. The outcome of the HRCT pattern with ground-glass attenuation, predominant reticulation, bronchiectasis, and accessory findings is less predictable.
In patients with anti-ARS autoantibodies, the imaging patterns suggestive of NSIP and organising pneumonia predominate, and consolidation is present in half of the cases that resolve most often [85]. ILD related to anti-PL12 autoantibodies is more frequently associated with reticulation and traction bronchiectasis than anti-Jo1 autoantibodies. As expected, patients with anti-Jo1 autoantibodies who experience severe ILD more frequently have a UIP pattern on imaging than those with less progressive disease (54% versus 18%, OR 5.02, 95% CI 1.05–243) [59], suggesting that the imaging pattern of ILD may have prognostic significance in this setting, similar to what is observed in idiopathic interstitial pneumonia.
Pulmonary function tests
ILD is typically associated with restrictive lung function impairment (total lung capacity (TLC) and diffusing capacity of the lung for carbon monoxide (DLCO) <80% predicted). DLCO is commonly reduced in patients with ILD, often at an earlier stage of the disease than TLC and forced vital capacity (FVC). Pulmonary function tests add important information to HRCT to evaluate severity, as they only partially correlate with HRCT scores and pattern. In addition, pulmonary function is more sensitive to changes upon therapy than HRCT abnormalities.
Interpretation of pulmonary function tests may be difficult in IIMs, because of the potential coexistence of ILD, respiratory muscle weakness and/or PAH, with all three impairing DLCO. Moreover, a restrictive pattern may be driven by ILD but also by respiratory muscle weakness, the evaluation of which is often difficult to interpret. Conversely, pseudo-stabilisation of a restrictive pattern may be spuriously attributed to improved diaphragmatic strength [51]. In one study, pulmonary function parameters improved early in the course of disease in some patients treated with a combination of high-dose corticosteroids and immunosuppressive agents, and even normalised in some patients despite persisting radiographic changes [80]. The clinical significance of persisting abnormalities on HRCT despite normal lung function is currently unclear. Clinicians must be aware of the possible discrepancy between imaging and lung function in patients with dermatomyositis- or polymyositis-ILD to interpret response to therapy and outcome.
BAL cellular profile
When combined with the results of a careful history, physical examination and thoracic imaging, analysis of the BAL differential cell count can contribute to the diagnosis of ILD. BAL differential cell count per se does not predict the pathology pattern, however, nor does it predict the response to therapy. Thus, the main role of BAL is to rule out infection. BAL is crucially important in patients with acute or subacute onset ILD, which may correspond to bacterial infection or to ILD related to IIM. It is even more relevant in subjects with progressive disease despite corticosteroid treatment, in order to diagnose rare occult infections including atypical mycobacteria, Pneumocystis jirovecii, Nocardia, intracellular bacteria, cytomegalovirus and fungi, especially in immunocompromised patients.
Attempts have been made to correlate BAL cellular patterns to imaging or histological patterns, or to the clinical presentation. For example, increased lymphocytes and eosinophils in the BAL were higher in dermatomyositis-ILD than in polymyositis-ILD in one study [86]. The CD4+/CD8+ ratio was greater in CADM than in those with classic dermatomyositis and rapidly progressive ILD [62, 70, 87]. Increased neutrophil counts in BAL were associated with a worse outcome in dermatomyositis- or polymyositis-ILD in two studies [47, 63]. However, BAL cellular patterns lack reproducibility and have no clear clinical relevance or treatment implications. In addition, the BAL cellular profile is subnormal or cannot be categorised in a large proportion of cases despite the presence of ILD [88].
Pathology
ILD can present with a variety of histological patterns in the setting of IIM. Although the available studies suffer from selection bias, histopathological patterns include in order of decreasing frequency NSIP (65%), UIP (15%), organising pneumonia (9%) and diffuse alveolar damage (8%) according to pooled data from six independent series (table 4) [13, 47, 79, 86, 89, 90], whereas lymphocytic interstitial pneumonia was present in only 1% of patients. The pattern of organising pneumonia may be underrepresented in these series, as clinicians may identify consolidation as a feature associated with a relatively good response to treatment and prognosis, and refer less frequently for surgical biopsy. Two patterns may coexist in various amounts in a patient, with for example mixed patterns of organising pneumonia and NSIP. The patterns found are very similar to those observed in the idiopathic setting, with subtle differences noted including fewer fibroblastic foci, smaller honeycombing spaces, higher germinal centres and total inflammation scores in UIP-associated with CTD than in idiopathic UIP (idiopathic pulmonary fibrosis) [91]. A correlation was reported in one study between pathology and response to treatment, with a good prognosis in cases of organising pneumonia, contrary to those with patterns of diffuse alveolar damage or UIP as expected [86].
Results of lung histopathology in patients with dermatomyositis- or polymyositis- associated interstitial lung disease
Overall, the pathology found in IIM-ILD is rather similar to that of idiopathic ILD. However, lung biopsy is now rarely performed in patients with CTD and ILD, as it generally does not alter management when compared with decisions based on less invasive procedures, e.g. clinical evaluation, pulmonary function tests, HRCT, and possibly BAL. A lung biopsy is discussed on an individual basis in rare cases with atypical imaging features, or when malignancy is considered.
Treatment
No randomised controlled trial has been performed to assess the efficacy and tolerance of corticosteroids and immunosuppressive drugs in patients with ILD associated with IIM. The efficacy of immunosuppressive drugs has mainly been reported in uncontrolled small retrospective series, which suggest a benefit of treatment but do not permit firm conclusions to be drawn. Thus, the indication for an immunosuppressive agent and the choice of the molecule are mostly based upon expert opinion, familiarity and experience of the clinician with a particular drug, and the tolerance profile. Treatments have to be initiated in cases of symptomatic or progressive ILD. Asymptomatic ILD must be carefully followed at diagnosis to determinate the disease evolution. Rapidly progressive ILD is poorly sensitive to corticosteroids alone and invariably requires combination with immunosuppressive therapies. Given a mortality rate close to 50%, the aim of therapy is to prevent progression to acute respiratory failure and death [51, 52].
Corticosteroids remain the cornerstone and first-line treatment of dermatomyositis- or polymyositis-associated ILD [13]. Oral regimens include 0.5–1 mg·kg−1 of prednisone daily for 3–8 weeks, followed by a gradual taper. In severe cases, one to three daily pulses of 1000 mg intravenous methylprednisolone are administrated [13]. Response to first-line corticosteroid therapy is considered adequate in 90–100% of patients with chronic ILD (table 5) [59, 63, 100], with much lower response rates (7–44%) in rapidly progressive ILD, which usually requires additional immunosuppressive therapy (table 6) [58, 62, 63]. In addition to refractory disease, immunosuppressive therapies may be used in cases of relapsing disease, or as steroid-sparing agents. A number of drugs have been used, including azathioprine, methotrexate, cyclophosphamide, cyclosporin A, tacrolimus, rituximab and i.v. immunoglobulins [13, 19, 47, 48, 58, 94]; however, no large studies have been published on their use in this context.
Studies reporting treatment effects (improvement/stabilisation) of corticosteroids and immunosuppressive drugs for chronic forms of DM- or PM-associated ILD
Studies reporting treatment effects (survival) of corticosteroids and immunosuppressive drugs in rapidly progressive ILD associated with DM or PM
The authors consider immunosuppressive therapy in addition to corticosteroids at diagnosis in patients with the most severe presentation, and in patients expected to poorly tolerate corticosteroids due to comorbidities, especially obesity. Other patients usually receive oral corticosteroids for the first 2–3 months; immunosuppressive agents may then be added to help reduce the dose of corticosteroids if tapering is difficult, and in those with an insufficient response to corticosteroids.
Azathioprine
Azathioprine, a derivative of 6-mercaptopurine agent, is an inhibitor of purine synthesis, thereby depleting cells of adenosine and guanosine nucleotides and inactivating T-cells, leading to immunosuppression [105]. It is usually administered orally at a dose of 2–3 mg·kg−1 daily. Azathioprine is widely used in subjects with anti-ARS autoantibodies and other forms of dermatomyositis/polymyositis with ILD, either as a corticosteroid-sparing agent or as maintenance therapy, solely or sometimes following induction with cyclophosphamide [59, 79, 102]. Published data suggest improvement of ILD in ∼75% of patients with chronic disease when it is used as first-line therapy (table 4) [59, 86, 92, 100, 102]. Whereas the results of azathioprine as second-line therapy were disappointing [102], in a very small series (seven patients) treatment with cyclophosphamide followed by azathioprine achieved remission in 58% of cases [59, 102], with better efficacy in cases with anti-ARS autoantibodies. Azathioprine has been used in very few patients with rapidly progressive ILD, but was unsuccessful (table 6) [58]. It should be kept in mind, however, that published retrospective data generally suffer from publication bias and may not be representative of the broader experience.
Methotrexate
Methotrexate is a folate antagonist, which inhibits dihydrofolate reductase. Because this enzyme is necessary in purine nucleotide synthesis, methotrexate administration results in decreased development and activation of T-lymphocytes. It is usually administered orally at a dose of 0.2–0.3 mg·kg−1 weekly. Case reports and retrospective studies have suggested that methotrexate may be effective in the treatment of chronic ILD, although the treatment effect was not detailed (table 5) [13, 79, 92]. Overall, experience with methotrexate is very limited in patients with IIM-ILD, and respiratory physicians often are reluctant to use this drug because of possible drug-induced pneumonitis.
Cyclosporin A
Cyclosporin A is a calcineurin inhibitor, which inhibits transcription of interleukin-2 and other cytokines [105], mainly targeting T-cells. Cyclosporin A can be prescribed as a single drug, or in combination with other immunosuppressive drugs in severe cases [58, 93, 97]. It seems to be effective as both a first- or second-line therapy in rapidly progressive ILD [103, 106]. In chronic ILD, an efficacy of 55% has been reported when used as a first-line treatment (table 5) [86, 92, 97, 100]; all patients responded to treatment when cyclosporin A was used as second-line in three small series [93, 102, 103]. In rapidly progressive ILD, cyclosporin A may be used as a first-line or second-line agent with response ranges of 56–80%, and 23–80%, respectively (table 5) [62, 58, 93, 96, 97, 100]. However, the use of cyclosporin A is limited by the endothelial and renal toxicity (which is potentially lethal) [103]. Moreover, published experience with cyclosporin A largely emanates from groups in Asia, with much more limited experience in European and American centres, and therefore the risk–benefit ratio and position of this drug in the treatment algorithm are still unclear.
Tacrolimus
Tacrolimus is a second-generation calcineurin inhibitor with greater potency compared with cyclosporin A. Several retrospective studies suggest its utility as second- [94] or third-line of treatment in chronic ILD (table 5) [93, 95]. Its use as a first-line treatment or in refractory cases of rapidly progressing ILD needs to be further explored, with anecdotal responses reported in four patients in three studies [62, 93, 94].
Mycophenolate mofetil
Mycophenolate mofetil, a prodrug of mycophenolic acid, is an inhibitor of inosine monophosphate dehydrogenase, which is the rate-limiting enzyme in de novo synthesis of guanosine nucleotides, controlling T- and B-lymphocyte development [107]. The remission or stabilisation of ILD was obtained in 80% of patients with chronic ILD when used as a first or second-line of treatment (table 5) [59, 101, 105, 108]. Due to its relatively favourable safety profile, mycophenolate mofetil is increasingly used in patients with dermatomyositis- or polymyositis-ILD who require an immunosuppressive drug in combination with corticosteroids.
Cyclosphosphamide
The alkylating agent cyclophosphamide, has been used to treat patients with dermatomyositis/polymyositis and rapidly progressive or chronic ILD, at a monthly dose of 0.6–0.7 mg·m−2 i.v. Due to the risk of secondary malignancies, cyclophosphamide should be restricted to cases of refractory ILD [96] or rapidly progressive ILD [62, 92]. The reported efficacy rate ranges from 38% to 100% in chronic ILD [59, 86, 96, 102] and rapidly progressive ILD (tables 5 and 6) [92].
Rituximab
Rituximab is a chimeric monoclonal antibody targeting the CD20 protein, which is primarily found on the cell membrane of B-lymphocytes. Rituximab may be effective in rapidly progressive ILD and in chronic ILD with anti-ARS autoantibodies. It is administered with two doses of 700 mg·m−2 or 1000 mg 2 weeks apart, or with four weekly infusions of 375 mg·m−2. Corticosteroids are often prescribed concomitantly to obtain remission. Maintenance therapy is administered for 3–6 months after induction with rituximab, consisting of azathioprine [97] or possibly cyclophosphamide [98, 109]. In seven patients with anti-ARS autoantibodies, a second infusion of 1000 mg was administered 6 months after induction as maintenance (table 6) [99]. However, experience with rituximab is still very limited in IIM-ILD and it cannot be considered a first-line therapy.
Association of immunosuppressive drugs or therapeutics
Some authors reported the use of two combined immunosuppressive drugs, such as mycophenolate mofetil plus cyclophosphamide, mycophenolate mofetil plus methotrexate, or cyclophosphamide plus tacrolimus, with a rationale of additive effects and reportedly good safety [110]. Some of the regimens do not use corticosteroids [110], which are nevertheless considered mandatory in the majority of cases.
Intravenous immunoglobulins and plasma exchange have been used in patients with life-threatening pulmonary involvement. Despite a reported benefit of immunoglobulins on the muscle involvement, these treatments seem to have very limited effects on rapidly progressive ILD [96, 104], and therefore are seldom used to treat the lung disease.
Outcomes
The mean mortality rate related to ILD is 14% [47]. Some series reported mortality as high as 45% in patients with dermatomyositis, with deaths mainly caused by ILD [86]; however, this probably does not represent the majority of patients. ILD is a major cause of death in JDM. The mortality rate of patients with chronic ILD related to CADM is similar to that of patients with ILD related to typical dermatomyositis [13, 51].
A prognostic score is dramatically lacking in the management of IIM-related ILD. The mean prognosis factor is the clinical presentation of dermatomyositis- or polymyositis-associated ILD. The rapidly progressive presentation of ILD can lead to acute respiratory failure with diffuse alveolar damage (similar to acute idiopathic interstitial pneumonia, formerly named Hamman–Rich syndrome) [47], for which the mortality is 50–75% [48, 58]. The course of myositis is similar in patients with or without ILD [47], although the prevalence of myositis refractory to corticosteroid therapy alone was higher in polymyositis/dermatomyositis patients with ILD than in those without ILD in one series, possibly because of a higher rate of anti-tRNA synthetase autoantibodies in ILD patients [47].
The prognosis can vary according to the presence of MSA. The risk of acute evolution of ILD and mortality are generally not increased in patients with anti-ARS antibodies compared with the other autoantibodies [52, 81, 96, 100]. Among patients with anti-ARS antibodies, the risk of pulmonary worsening and death seems to be similar in those with anti-Jo1 and non-Jo1 antibodies, but conclusions diverge according to the study [4, 8, 15, 111]. Prognosis of ILD associated with anti-PM/Scl autoantibodies is similar to other dermatomyositis- or polymyositis-associated ILD [19, 32]. Anti-PM/Scl patients have symptomatic pulmonary involvement with a globally favourable prognosis comparable to that of patients with anti-ARS autoantibodies [19]. Mortality from ILD is low in patients with anti-PM/Scl autoantibodies, ranging from 0% to 11% in two small retrospective cohorts [19, 32]. The survival rate of ILD associated with anti-MDA5 autoantibodies is lower than that of anti-ARS ILD [56].
Pulmonary arterial hypertension
Epidemiology
Precapillary pulmonary hypertension associated with dermatomyositis was first described in 1956 [112], and was reported more recently in case reports [113, 114] and short series [5]. The estimated frequency of pulmonary hypertension is 8% in patients with anti-ARS autoantibodies [5], and may be as high as 29% in patients with anti-PL7 autoantibodies [115]. Pulmonary hypertension can precede the diagnosis of myositis [116]; however, it is related to ILD (pulmonary hypertension group 3) in most, if not all, cases often after several years of disease duration [5]. These patients frequently experience clinical signs of SSc such as sclerodactyly, skin sclerosis, or digital ulcers [5]. In one series, arthralgia were less frequent at diagnosis in patients who developed pre-capillary pulmonary hypertension than in those who did not (38% versus 65%) [5].
Pathogenesis and pathology
Isolated pulmonary hypertension is very rare in dermatomyositis/polymyositis in the absence of ILD. Chronic hypoxic pulmonary vasoconstriction is one major factor driving the onset of pulmonary hypertension in patients with IIM-ILD; however, involvement of pulmonary vessels by the diffuse infiltrative and inflammatory process is likely to contribute to pathogenesis, as well as other less identified factors [5, 117]. In fact, pulmonary hypertension exists in the absence of significant hypoxia secondary to progressive ILD [5, 117]. Cutaneous features of SSc including Raynaud's phenomenon, digital ulcers, sclerodactyly and puffy fingers are frequently reported in patients with anti-ARS autoantibodies, and are more frequent in overt pulmonary hypertension [8, 118]. Two case reports in overt and hypomyopathic polymyositis describe severe right ventricular hypertrophy and plexogenic severe intimal fibrosis consistent with pulmonary arteriopathy at autopsy [113, 117]. Obliterative pulmonary vasculopathy (resulting from proliferation of endothelial and vascular smooth muscle cells) similar to that of SSc, but involving more proximal pulmonary arterioles, often coexists [117]. As in other forms of severe PAH, vascular remodelling involves all three layers of the pulmonary artery wall, with intimal thickening and medial hypertrophy, thus resembling changes seen in idiopathic PAH. The role in pathogenesis of the mechanisms described in SSc, ILD and pulmonary hypertension, namely vascular inflammation, perivascular fibrosis and thrombotic angiopathy, is unknown in IIM [119]. Similarly, the implication of growth factors (transforming growth factor, vascular endothelial growth factor, basic fibroblast growth factor, platelet-derived growth factor and interleukin-6) and the relative contributions of the endothelium-derived vasoactive factors (endothelin, nitric oxide and prostanoids) have not been explored in the pathogenesis of pulmonary hypertension associated with anti-ARS autoantibodies.
Treatment
Nonspecific treatments for pulmonary hypertension including phosphodiesterase-5 inhibitors (sildenafil and tadalafil), endothelin receptor antagonists (bosentan and ambrisentan) and prostacyclin derivatives (i.v. epoprostenol, subcutaneous treprostinil and nebulised iloprost) may be considered in patients with dermatomyositis- or polymyositis-associated with pulmonary hypertension [120–122]. Treatment should be reserved, however, to those rare patients with group 1 PAH with no significant ventilatory impairment resulting from ILD, whereas treatment is not recommended in those with group 3 pulmonary hypertension considered secondary to ILD [119]. It is unknown whether immunosuppressive therapies and corticosteroids (which may improve pulmonary hypertension in patients with systemic lupus) may be beneficial in patients with IIM and pulmonary hypertension [123, 124].
Outcome
Presence of both ILD and pulmonary hypertension is associated with dramatically low long-term survival, with a 3-year survival rate of 58% [5]. The prognosis of isolated pulmonary hypertension in dermatomyositis/polymyositis seems to be better, with a 3-year survival of 100% reported in seven patients [125]; however, experience is very limited.
Combined pulmonary fibrosis and emphysema
CPFE is a recently identified syndrome in smokers or ex-smokers characterised by dyspnoea that is often severe, preserved lung volumes, severely impaired gas exchange and an increased risk of pulmonary hypertension associated with a dismal prognosis [126, 127], and possibly lung cancer [128]. The diagnosis is based on the presence of both emphysema predominating in the upper lobes, and frequent paraseptal and ILD findings in the lower lung zones with typical velcro crackles at auscultation. Patients with CPFE experience a slower decline in FVC and DLCO than ILD patients without emphysema, and thus changes in these parameters alone may not be reliable indicators of disease progression [126, 127].
In a series of 34 patients with CTD, one patient was diagnosed as polymyositis, and another patient with myalgia, elevated creatine phosphokinase levels and Raynaud's phenomenon had anti-Jo1 autoantibodies. Patients with CTD-associated CPFE were younger and more frequently female compared with those with “idiopathic” (smoking-related) CPFE [129], with a similar prognosis. Patients with CTD-associated CPFE were more likely to be male and to be smokers than patients with CTD-associated ILD without emphysema [129], with no survival difference. Little is known about the prognosis and treatment of CPFE in the setting of dermatomyositis/polymyositis. Cessation of smoking is imperative.
Pleural involvement
Pneumomediastinum
Spontaneous pneumomediastinum has been reported as a rare complication of various types of ILD, especially ILD associated with dermatomyositis/polymyositis [3]. This event precedes the diagnosis of CTD in 18% of cases [3]. 70 cases, including two JDM [130, 131], were described, mainly secondary to dermatomyositis (95%) [3, 130–137]. ∼6–8% of patients with polymyositis/dermatomyositis and ILD may develop a pneumomediastinum, leading to death in 35–66% of cases [3, 60, 138]. The presentation of pneumomediastinum ranges from acute respiratory failure to fortuitous diagnosis (33% of cases) [3, 139]. The predominant manifestations at onset are increased dyspnoea, cervical or facial swelling, subcutaneous emphysema, cervical pain and cough [3, 140]. Pneumothorax coexists in 40% of cases [3]. Pneumomediastinum is sometimes complicated by asymptomatic pneumorrachis, i.e. epidural emphysema, diagnosed by HRCT [139]. Relapse of pneumodiastinum occurs in a quarter of cases [3].
Pathogenesis and pathology
In the setting of ILD, pneumomediastinum usually results from rupture of the subpleural or paracardial blebs due to the distortion of the lung architecture [3]. However, these abnormalities may have been underreported. The development of pneumothorax, pneumomediastinum, or both probably depends on the location of the rupture (peripheral for pneumothorax and adjacent to vessels for pneumomediastinum) [3]. An increase in intra-alveolar pressure during air travel [132] may have been a triggering factor in 45% of cases [3]. Kono et al. [138] hypothesised a relationship between the occurrence of pneumomediastinum and dermatomyositis-associated vasculitis leading to the necrosis of the bronchial wall. Patients with low vital capacity (VC) before pneumomediastinum are at higher risk of death [3].
Treatment
The treatment of pneumomediastinum consists of bed rest, thoracic drainage of the associated pneumothorax, and control of ILD by drug therapy, including corticosteroids in all cases, and occasionally methotrexate, i.v. immunoglobulins, cyclosporin A, azathioprine, mycophenolate mofetil, or rituximab [3, 130–137, 141].
Pneumothorax
Recurrent spontaneous pneumothorax was reported in a 17-year-old man diagnosed with JPM, in the absence of ILD [130]. Intra-operative observation and histopathological study demonstrated numerous bleb-like lesions below the visceral pleura. In other reported cases, pneumothorax was invariably secondary to ILD [97].
Pleural effusion
Exudative pleural effusion is a rare manifestation of dermatomyositis or polymyositis, and has mainly been described in case reports. In a series of dermatomyositis- or polymyositis-related ILD the prevalence was estimated as 5% [86]. It may be isolated or associated with ILD or pericardial effusion [142–144]. A CTD-specific myocarditis with congestive failure must be ruled out in cases of transudate effusion [144].
Indirect pulmonary complications
Infection
Infectious complications in dermatomyositis/polymyositis have been described in up to 37% of patients, with aspiration pneumonia being the most frequent infection. Patients receiving immunosuppressive drugs, or having oesophageal involvement, ventilatory insufficiency, cancer or myalgia are at higher risk of infections [145].
Pyogenic infection
Lungs are reported as the most common site of severe pyogenic infection in dermatomyositis/polymyositis patients (70% of cases) [17, 59, 86]. Aspiration pneumonia is a critical event in polymyositis/dermatomyositis patients with a prevalence of 7–20%, mainly within the first year after diagnosis [59, 60]. Most patients with aspiration pneumonia exhibited concomitant oesophageal involvement and ventilatory insufficiency related to respiratory striated muscle weakness [17].
Opportunistic infection
Pulmonary tuberculosis occurred in one (0.7%) patient receiving steroids in a series of 279 patients [17]. Pulmonary infection by Mycobacterium xenopi was reported in a patient treated with corticosteroids, and infection by Mycobacterium peregrinum in a patient taking corticosteroids and infliximab [17]. Other opportunistic lung infections secondary to corticosteroids and various immunosuppressive drugs described in dermatomyositis/polymyositis patients include pneumonia or pleural invasion by Nocardia spp. (seven cases) [146–148], pneumonia secondary to Candida albicans (one case) [145], Aspergillus fumigatus (three cases) [17, 149, 150], Aspergillus niger (one case) [150], lung abscess due to Cryptococcus laurentii (one case) [151], blastomycosis (one case) [152] and cytomegalovirus (four cases) [145, 153, 154]. Pneumocystis jirovecii infection was described after administration of prednisone, azathioprine, methotrexate, co-administration of methotrexate and azathioprine, and rituximab [17, 98].
Drug toxicity
Representative drugs that have been reported to cause drug-induced ILD in dermatomyositis/polymyositis are listed in table 6 [155]. An exhaustive list of medications with their reported pulmonary toxicities can be found online (www.pneumotox.com). Drug-induced ILD should be differentiated from diffuse lung diseases, including idiopathic ILD, ILD associated with CTD, acute and chronic hypersensitivity pneumonia, eosinophilic pneumonia, acute lung injury, acute respiratory distress syndrome, and Pneumocystis pneumonia. However, it is particularly difficult to determine whether new onset opacity at imaging is attributable to the underlying disease or to drug-induced ILD, as the signs and symptoms, HRCT pattern and biopsy findings are nonspecific. BAL may prove useful to identify microorganisms in culture, or to demonstrate increased eosinophil count or mixed lymphocytic and neutrophilic cellular patterns compatible with drug-induced ILD. The temporal relationship between the new/superimposed pulmonary manifestation and the initiation of the suspected medication may help to distinguish specific ILD related to IIM from drug-induced manifestations.
Malignancy
The association between IIM and cancer has been extensively studied in adults [1]. Among all malignancies, lung cancer is one of the leading causes of paraneoplastic forms of dermatomyositis/polymyositis, with a prevalence ranging from 1% to 5% in large series [156–159]. Conversely, paraneoplastic forms are extremely rare in JDM, contrasting with the relatively frequent malignancy observed in adults with dermatomyositis.
Dermatomyositis and polymyositis are significantly associated with a four-fold increase in the risk of malignancy compared with the general population [156]. The risk is maximal within the first year of follow-up and in Asian populations, and is higher in cases of severe dermatomyositis rash suggestive of intense vasculitis (cutaneous ulcers and periungual erythema), older age, male sex, higher C-reactive protein, elevated erythrocyte sedimentation rate and dysphagia [156, 160, 161]. Conversely, the frequency of cancer is lower among patients with anti-extractable nuclear antigen autoantibodies, anti-Jo1 autoantibodies and corresponding clinical features (ILD, Raynaud's phenomenon and arthralgia) [47, 160], but these findings cannot allow CAM to be ruled out [63].
In 2006, a new myositis-specific autoantibody (anti-p155) directed against transcriptional intermediary factor 1γ (TIF1γ) was described in dermatomyositis patients as a good marker of CAM, with an inverse (protective) association with ILD (table 2) [162]. Kasuya et al. [163] described the presence of TIF1γ-bearing neoplasm in anti-p155/140 antibody-positive dermatomyositis and hypothesised the antigenic role of overexpressed TIF1γ for the induction of paraneoplastic dermatomyositis. The clinical phenotype associated with these autoantibodies encompasses rashes evocative of typical dermatomyositis as a first manifestation, including periungual changes, shawl and/or V-signs at diagnosis. The rashes persist despite the remission of myositis [11]. A meta-analysis of 312 adults from six studies reported that anti-TIF1γ autoantibodies may be linked with cancer in dermatomyositis patients with a pooled sensitivity and specificity of anti-TIF1γ for diagnosing CAM of 78% and 89%, respectively [164]. Anti-TIF1γ had positive and negative predictive values of 58% and 95%, respectively.
Patients with CAM have a worse prognosis when compared with patients with idiopathic polymyositis or dermatomyositis. The mortality rate in this group of subjects is high and their life expectancy is related to the cancer and not to myositis itself. Since older age is a well-established risk factor for CAM, a thorough search for cancer should be performed in elderly patients with IIM.
Respiratory muscle weakness
Respiratory muscle involvement in IIM is extremely rare. The prevalence of respiratory failure due to respiratory muscle weakness is still unknown in this setting. The involvements of the respiratory muscles and the limb muscles are sometimes dissociated [165]. Patients suspected of having respiratory muscle involvement should be identified promptly because it is potentially life threatening [166].
In cases with moderate involvement, the standard pulmonary function tests may be unremarkable [167]. With progressive disease, restrictive physiology can be observed, with low TLC and VC, with normal FVC and DLCO [167]. Maximal inspiratory (and expiratory) pressures are decreased, as well as the sniff test (measurement of inspiratory pressure during a sniff). Supine respiratory function tests are useful for clinical diagnosis and respiratory risk stratification [167].
Patients with symptoms suggestive of respiratory muscle involvement should be closely monitored in hospital with a daily assessment of FVC using a bedside spirometer rather than peak flow [166]. Patients with FVC <15 mL·kg−1 should be referred to an intensive care unit for monitoring and possible noninvasive mechanical ventilation [166]. The other critical predictive parameters include maximal inspiratory and expiratory pressures <30% predicted, and VC <55% predicted [167].
The treatment of respiratory weakness does not differ from that of other muscular involvement. In a retrospective series of 18 patients with myopathic restrictive respiratory involvement treated by corticosteroids or immunosuppressive drugs including azathioprine and cyclosporin A, complete remission was reported with therapy in 83% [48]. Two patients were treated with home mechanical ventilation [48]. The role of i.v. immunoglobulins in this condition is unclear.
Cardiac involvement
Oppenheim and colleagues first reported cardiac involvement in dermatomyositis in 1899 [168]. The prevalence of cardiac involvement in dermatomyositis/polymyositis varies between 9% and 72% according to the series and method of detection [169–173]. Clinically significant heart involvement is found in 10–15% of patients [174, 175]. Congestive heart failure, left ventricular diastolic dysfunction, coronary artery disease and arrhythmias may contribute to dyspnoea [167, 168, 176]. Pulmonary oedema due to myocarditis and/or cardiomyopathy usually occurs together with active muscular disease. Subclinical cardiac manifestations of myositis include various conduction blocks, and infra- or supraventricular arrhythmias in 30% of patients [167, 173]. The incidence of pericardial effusion is 15% [177].
Heart involvement is an independent factor of mortality in dermatomyositis/polymyositis [174]. Deaths related to myocarditis occurred in 1–2% of patients in a recent series of dermatomyositis/polymyositis [174]. Besides immunoppressive therapy, cardiotropic drugs including β-blockers and angiotensin-converting enzyme inhibitors may also be of benefit to patients. Atrioventricular block has been successfully treated using pacemakers [173].
Summary
ILD is a leading cause of morbidity in dermatomyositis/polymyositis, especially rapidly progressive ILD. New autoantibodies associated with various clinical phenotypes have facilitated the diagnosis of IIMs, and contributed to better estimates of the risk and prognosis of ILD. Treatment depends on the severity and progression of ILD at diagnosis, and includes high-dose corticosteroids, often with additional immunosuppressive drugs (including azathioprine, mycophenolate mofetil or methotrexate in chronic ILD; and cyclosporin A or i.v. cyclophosphamide in rapidly progressive ILD). Further studies are needed to better understand the pathophysiology of ILD related to dermatomyositis/polymyositis, and to investigate the effectiveness and tolerance of targeted therapies in randomised controlled trials.
Footnotes
This version of this article has been amended. The amendment is outlined in the correction published in the September 2015 issue of the European Respiratory Review [DOI: 10.1183/16000617.50002015].
Conflict of interest: Disclosures can be found alongside the online version of this article at err.ersjournals.com
Provenance: Submitted article, peer reviewed.
- Received March 11, 2015.
- Accepted April 22, 2015.
- Copyright ©ERS 2015.
ERR articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References









