





Abstract
Over the past 10–15 years, the diagnosis of α1-antitrypsin deficiency (AATD) has markedly improved as a result of increasing awareness and the publication of diagnostic recommendations by the American Thoracic Society (ATS)/European Respiratory Society (ERS). Nevertheless, the condition remains substantially underdiagnosed. Furthermore, when AATD is diagnosed there is a delay before treatment is introduced. This may help explain why AATD is the fourth most common cause of lung transplantation. Clearly we need to do better. The ATS/ERS recommend testing high-risk groups, such as: all chronic obstructive pulmonary disease patients; all nonresponsive asthmatic adults/adolescents; all cases of cryptogenic cirrhosis/liver disease; subjects with granulomatosis with polyangitis; bronchiectasis of unknown aetiology; panniculitis and first-degree relatives of patients with AATD. In terms of laboratory diagnosis, measurement of α1-antitrypsin levels will identify patients with protein deficiency, but cannot differentiate between the various genetic subtypes of AATD. Phenotyping is the current gold standard for detecting rare variants of AATD (except null variants), while advances in molecular diagnostics are making genotyping more effective. An accurate diagnosis facilitates the physician's ability to actively intervene with measures such as smoking cessation and perhaps augmentation therapy, and it will also help provide a better understanding of the natural history of the disease.
Abstract
Due to advances in testing and increased awareness, AATD is now a relatively common, rarely diagnosed condition http://ow.ly/HeJUr
Introduction
Despite the fact that α1-antitrypsin deficiency (AATD) is commonly labelled a rare disease, it is one of the most common autosomal genetic disorders in humans. Clinically, AATD is associated with an increased risk for developing chronic obstructive pulmonary disease (COPD). The 2014 Global Initiative for Chronic Obstructive Lung Disease recommendations [1] quote the World Health Organization, who recommend that COPD patients from areas with a particularly high prevalence of AATD should be screened for the genetic disorder [2]. They also note that compared to other forms of COPD, typical patients with AATD tend to present at a younger age (<45 years) with lower lobe emphysema and suggest that family members can be identified. They describe a serum concentration of α1-antitrypsin (AAT) <15–20% of normal as highly suggestive of homozygous AATD [2]. In contrast, the recommendations from the American Thoracic Society (ATS)/European Respiratory Society (ERS) guidelines are more encompassing, recommending the testing of: all COPD patients; all nonresponsive asthmatic adults/adolescents; all people with cryptogenic cirrhosis/liver disease; granulomatosis with polyangitis; bronchiectasis of unknown aetiology; panniculitis and possibly other skin disorders; and first-degree relatives of patients/carriers with AATD [3].
The epidemiology of AATD has been widely investigated over the past 15–20 years following publication of seminal papers by Hutchison [4], de Serres [5] and Luisetti and Seersholm [6]. However, there are still gaps in our knowledge in terms of the prevalence of the more common AAT deficient variants PI*S and PI*Z, particularly in Eastern Europe, South America, East Asia and developing countries [6]. Moreover, the available data may not be as accurate as we would like. For example, Luisetti and Seersholm [6] presented allele frequencies for PI*S and PI*Z of 0.02–0.04 and 0.005–0.015, respectively, in Ireland, whereas much higher rates were recorded in a study of 1100 randomly sampled individuals from the Trinity College Biobank (Dublin, Ireland) with frequencies for PI*S and PI*Z of 0.054 and 0.022, respectively [7]. Excluding newborn screening studies, this is one of the few truly representative investigations of AAT allele frequencies in a general population as the subjects were randomly selected from the electoral register. In the past, studies have been biased to varying degrees as they interrogated cohorts of symptomatic individuals, soldiers, sailors, hospital staff, pregnant women, individuals with rheumatic disorders or other diseases, newborn infants and employees. Studies of blood bank donors also risk underestimating AATD as these donors tend to be healthy individuals. Consequently, there is a lack of accurate, reliable data on the prevalence of PI*S and PI*Z worldwide and even less is understood about the genetic epidemiology of rare AAT deficient variants [6].
Screening studies have estimated that <10% of affected individuals are adequately diagnosed [8]. Whilst this is an improvement from earlier studies in which <5% of expected cases were identified [6], the rate remains unacceptably low. Moreover, there is evidence that among individuals who have been correctly diagnosed, treatment may be delayed for between 5 and 7 years, and it can be reasonably assumed that during this period lung function will decline [9, 10]. This may help explain data presented by the International Society of Heart and Lung transplantation which identified AATD as the fourth most common reason for lung transplantation, accounting for ∼6% of cases (table 1) [11]. This is a high percentage for a rare disease and highlights the need for better diagnosis and testing.
Indications for adult lung transplants between January 1995 and June 2012
Different types of testing for patients with genetic disorders
There are three approaches to the diagnosis of AATD: 1) diagnostic testing of individuals with symptoms/signs consistent with AAT-related disease; 2) predispositional testing of individuals who may be at high-risk of having AATD; and 3) screening.
Diagnostic and predispositional testing
In the past, diagnostic testing of all persons with signs and symptoms of AATD generally meant testing of individuals with early onset, primarily lower lobe, emphysema. This paradigm has not served us well and has led to under diagnosis and late diagnosis. Now, testing is in line with the ATS/ERS guidelines [3], which has led to a significantly increased detection rate. Predispositional testing generally involves follow-up of asymptomatic subjects in whom a gene mutation has been identified, usually family members with low AAT levels. While development of disease related to AATD is likely in the future for these individuals, it is not certain.
To address a lack of data relating to AATD in Ireland a study was undertaken to analyse 1100 individuals from the general population (from the Trinity College Biobank collection) and a targeted group as described by the ATS/ERS guidelines [7]. It was intended to compare whether targeted investigation increased the level of diagnosis across all AATD allele groups. In the targeted group, ∼50% were tested because of a diagnosis of COPD, with the remainder of testing attributed to low levels of AAT (16%), family history (14%), liver disease (9%) and asthma (9%) [7]. In the general population sample represented by the Trinity College Biobank, the frequencies of PI*S and PI*Z mutations were much higher than previously estimated and were amongst some of the highest reported. The targeting of specific populations in the detection programme resulted in a significant enrichment of the numbers of patients identified with some form of AAT deficiency. In particular, the high frequency of the PI*Z allele in the targeted population underlies the role of this mutation in the pathogenesis of AATD. Interestingly, PI*S allele levels were similar in the targeted and general population groups. This study emphasised the importance of early diagnosis so that appropriate medical follow-up and lifestyle changes can be instituted to help prevent or delay the development of the lung and/or liver disease associated with AATD [7]. Following on from this study, the Irish Targeted Detection Programme continues to receive >200 samples per month from over 30 hospitals or general practitioner clinics nationwide and we have now instituted a red-flag system for hospital laboratories so that patients with a low level of AAT (<1 g·L−1) are automatically specifically screened for AATD.
Screening
The ATS/ERS guidelines state that neonatal screening for AAT is not recommended and pointed out that while a Swedish study showed that neonatal screening reduced smoking rates following detection, with no adverse psychological effects [3, 12], it was associated with an increase in parental distress and had a negative impact on the mother–child relationship [12]. The ATS/ERS guidelines do not generally recommend screening in adolescents aged >11 years, but suggest that screening should be discussed with individuals in areas with a high prevalence of AATD or if smoking rates are high, providing that adequate counselling is given. Recommendations for adults are similar to those for adolescents [3]. At present, screening guidelines are evolving and appear to be quite dynamic with a number of important questions needing to be answered. For example, should we try to identify all AAT variants or just PI*ZZ mutations, or those associated with diseases of the lungs, liver, and skin? What about PI*MZ and PI*SZ variants, and rarer mutations?
Severe AATD results in an increased risk of COPD and typically occurs in PI*ZZ homozygous individuals. However, new data suggest that some patients with heterozygous PI*MZ may be at increased risk for airflow obstruction particularly if they smoke [13]. For many years PI*MZ individuals were classified as carriers which suggested that they did not suffer from active disease. A recent family-based study determined the risk of COPD in PI*MZ and PI*MM individuals [13]. Some of the key findings are presented in table 2. In nonsmokers, the results for the two groups were comparable, but for smokers forced expiratory volume in 1 s (FEV1)/forced vital capacity (FVC), FEV1 % predicted and forced expiratory flow at 25–75% of FVC were all reduced in the PI*MZ group. The results indicate that PI*MZ ever-smokers have an increased risk of developing COPD compared with PI*MM individuals who were ever-smokers.
Results from a family-based study to determine the risk of chronic obstructive pulmonary disease in PI*MM and PI*MZ individuals
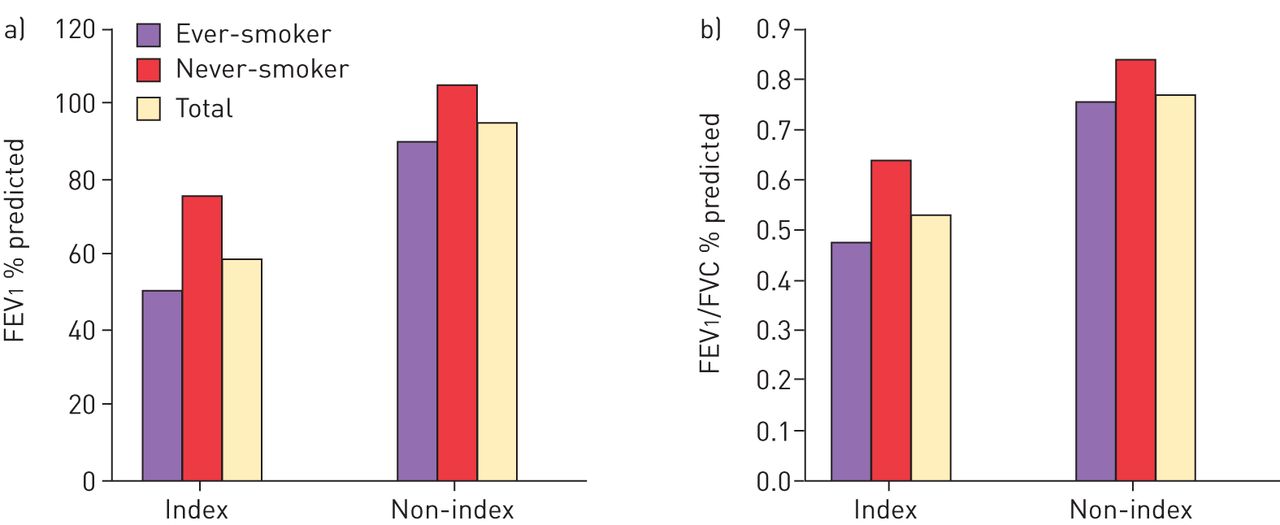
The role of heterozygous PI*SZ in disease is also uncertain and was investigated by Seersholm and Kok-Jensen [14] using the Danish AATD registry which contains details of patients with respiratory symptoms (index cases: n=28), as well as subjects included because of family history who were not necessarily symptomatic (non-index cases: n=66). Demographically, the index and non-index groups were very similar with the exception that there were more current smokers in the non-index group (33% versus 21%) and more former smokers in the index group (39% versus 14%). There were eight deaths in the index group (pulmonary emphysema: n=6; pulmonary fibrosis: n=1; colon cancer: n=1) and five deaths in the non-index group (pulmonary emphysema: n=2, pneumonia: n=2; cerebral haemorrhage: n=1). The mean FEV1 % predicted and FEV1/FVC ratio findings in these populations are shown in figure 1. Respiratory function was impaired in the index cases compared with the non-index cases irrespective of whether the patients were ever- or never-smokers. In addition, in the index and non-index groups, both FEV1 % predicted and FEV1/FVC ratio were lower in ever-smokers compared with never-smokers. Based on an analysis of the non-index group the authors concluded that it appears that only a small proportion of PI*SZ individuals are at increased risk of developing pulmonary emphysema, and it occurs at an older age than in PI*ZZ patients [14]. Additionally, PI*SZ patients with active disease (index cases with decreased FEV1 % predicted and symptoms) have reduced survival compared with the general population [14].

a) Forced expiratory volume in 1 s (FEV1) and b) FEV1/forced vital capacity (FVC) ratio in index and non-index patients with PI*SZ α1-antitrypsin deficiency included in the Danish α1-antitrypsin deficiency register. Data from [14].
In a report by the Alpha 1-Antitrypsin Deficiency Registry Study Group, the clinical characteristics of 59 individuals with the PI*SZ phenotype and AATD were investigated [15]. At the time of the report there were 1129 individuals with AAT levels ≤11 µM and the findings for the PI*SZ patients were compared with data from PI*ZZ patients. In nonsmokers, airflow obstruction, as estimated by FEV1 <80% predicted, occurred less frequently in the PI*SZ group compared with the PI*ZZ group. One of the most striking findings in this analysis involved ex- and current smokers. In these patients most PI*SZ individuals with an FEV1 <80% were ex- or current smokers while few PI*ZZ smokers had an FEV1 in the normal or near normal range. The authors concluded that in smokers the PI*SZ phenotype confers a significant risk of the development of COPD and in nonsmokers there may be no such added risk [15].
What about rarer AATD variants? In Ireland we have screened >12 000 patients and found that >29% have at least one abnormal AAT allele and ∼1.4% have a rare mutation (table 3). It is important that we are able to identify these rarer variants since they may be associated with lung or lung and liver disease [7, 16].
Rare α1-antitrypsin variants detected during screening of >12 000 patients in Ireland
Laboratory diagnosis of AATD
As part of the AATD screening programme in Ireland two principal tests are performed to assist with diagnosis. First, AAT levels are measured in serum or plasma (and this can be done in most hospitals) and a cut-off of 1 g·L−1 has been set. Secondly, samples with an AAT level <1 g·L−1 are sent for qualitative isoelectric focusing electrophoresis with immunoblotting. Using this method we can easily differentiate between the AATD phenotypes (fig. 2). By analysing the various bands obtained during isoelectric focussing electrophoresis (e.g. I, F, S, Z and Zbristol) we were able to identify the majority of rarer mutations associated with AATD [7]. However, there are some caveats that need to be addressed. If an MM variant is identified and the level is <0.9 g·L−1, or an MZ/MS variant is identified with a level <0.7 g L−1, then DNA sequencing should be performed. If an SS or ZZ phenotype is found then genotyping should be performed to exclude the possibility of a null mutation.

{kind=link}
{kind=link}
a) Isoelectric focusing electrophoresis of a rare Mmalton variant (homozygous) showing unusual M-like and Z-like bands, which was confirmed by b) DNA sequencing. Data from the Irish National Targeted Detection Programme (courtesy of N.G. McElvaney).
Case example: AATD rare variant
A patient presented with significant lung disease and a low level of AAT (0.19 g·L−1) on blood testing. Isoelectric focusing and immunoblotting showed unusual M-like bands and Z-like bands (fig. 2) and the patient was classified MM variant using a commercial genotype assay. These lines of evidence led us to conclude the presence of two rare mutations and a sample was sent for DNA sequencing which identified a very rare Mmalton variant in homozygosity (fig. 2). This was an important diagnosis to make since AATD resulting from a Mmalton variant is usually associated with an increased risk of both lung and liver disease.
What is the optimal AAT level cut-off point?
The optimal cut-off point for AAT levels to detect AATD or its genetic causes has been the subject of scientific debate and the answer depends on the aims of the investigation. Based upon the findings of Ferrarotti et al. [17], the following cut-off points have been suggested. 1) If the aim is to identify genotypes with increased risk for emphysema, a cut-off point of 1 g·L−1 (95.8% sensitivity, 94.8% specificity) has proven satisfactory. 2) However, if the goal is to ensure all S or Z alleles are detected then a cut-off point of 1.1 g·L−1 (73.4% sensitivity, 88.5% specificity) is more suitable. 3) In other studies a cut-off of 1.13 g·L−1 has been employed since no Z alleles (or PI*SS) have been found above this level (100% sensitivity, 78% specificity; for AATD, 79% sensitivity, 83% specificity if related to S or Z mutations). Above this level there is a marked increase in the number of false positives.
Recently, genotyping using PCR methodologies has moved forward and can now detect S, Z and F variants and analytical genotyping kits are becoming available that can measure ∼25 AATD alleles. Next generation diagnostic kits will soon be available that can measure most, if not all, AATD mutations.
Conclusion
In patients with AATD it is important to detect deficiency alleles. The steps performed in Ireland to ensure that this is the case are as follows: 1) serum/plasma AAT levels are measured by means of immune turbidimetry, cut-off: 1 g·L−1; 2) phenotyping of those samples with AAT levels lower than the cut-off; 3) SS or ZZ phenotypes undergo genotyping to exclude the possibility of a null mutation; 4) MM variants with AAT levels <0.9 g·L−1 undergo DNA sequencing; and 5) MZ/MS variants with levels <0.7 g L−1 also undergo DNA sequencing. This will cover most eventualities but not all, the null (isola di procida) is a classic example.
The importance of an accurate diagnosis is that it facilitates our ability to actively intervene with measures such as smoking cessation and perhaps augmentation therapy, and it also helps us learn more about the natural history of the disease. There is a lot we still need to know about AATD. We have a greater understanding of the role of AATD in emphysema and COPD, but we have much more to learn about its role in liver and skin disorders. The more we understand about the interaction between S, Z and rare mutations the better equipped we will be to implement appropriate disease management strategies to defeat disease progression in this debilitating condition.
Acknowledgements
Writing assistance was provided by Content Ed Net (Madrid, Spain) with funding from Grifols (Barcelona, Spain).
Footnotes
Conflict of interest: Disclosures can be found alongside the online version of this article at err.ersjournals.com
Provenance: Publication of this peer-reviewed article was supported by Grifols, Barcelona, Spain (article sponsor, European Respiratory Review issue 135).
- Received November 13, 2014.
- Accepted December 20, 2014.
- Copyright ©ERS 2015.
ERR articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References









