





Abstract
Chronic infection and inflammation are defining characteristics of cystic fibrosis (CF) airway disease. Conditions within the airways of patients living with CF are conducive to colonisation by a variety of opportunistic bacterial, viral and fungal pathogens. Improved molecular identification of microorganisms has begun to emphasise the polymicrobial nature of infections in the CF airway microenvironment. Changes to CF airway physiology through loss of cystic fibrosis transmembrane conductance regulator functionality result in a wide range of immune dysfunctions, which permit pathogen colonisation and persistence. This review will summarise the current understanding of how CF pathogens infect, interact with and evade the CF host.
Introduction
Cystic fibrosis (CF) is an autosomal recessive disorder arising from functional deficiency of the cystic fibrosis transmembrane conductance regulator (CFTR). Although this channel is primarily responsible for facilitating conductance of chloride ions across membranes, it also transports other molecules including bicarbonate [1, 2], glutathione [3, 4], and thiocyanate [5]. The loss of CFTR function has wide-ranging effects on human health, with disease manifestation in the pancreas, liver, gastrointestinal tract and lungs, as well as in the male reproductive tract. Lung disease is responsible for the majority of morbidity and death in CF and is the largest obstacle in therapy. CF lung disease is characterised by high levels of infiltrating cells (especially neutrophils, but also lymphocytes and macrophages [6]), elevated levels of inflammatory cytokines [7, 8], and chronic colonisation by opportunistic pathogens [9]. These microorganisms exploit niches in the CF lung environment to elicit sustained inflammatory responses, resulting in significant structural damage to the airways and decline of pulmonary function [10]. Understanding how microorganisms colonise and persist in the lung environment while subverting the host immune response is a critical step in developing specifically targeted anti-inflammatories that can be used in conjunction with antibiotic and airway clearance therapies. Loss of CFTR can be understood to affect host–pathogen interactions through several key functions: alteration of the lung microenvironment and permissibility of infection, endogenous dysregulation of host defence mechanisms, and by directly affecting pathogen recognition.
The CF lung microenvironment
The mechanism through which the loss of Cl- secretion through the CFTR results in the decline of the mucus and periciliary layer volumes of the airway surface liquid (ASL) is still unclear [11]. One of the leading hypotheses argues that the CFTR may serve to regulate Na+ and, consequently, water reabsorption by epithelial Na+ channels (ENaC) expressed on the airway epithelium. This has been supported by studies showing that overexpression of ENaC results in airway disease that is characteristic of CF in mice [12] and various cellular models showing that ENaC expression and function is regulated by CFTR [13, 14]. This has been contested in studies that have found that Na+ conductance is unaffected in CF airway epithelia of pigs [15] and humans [16] and that the CF disease phenotype may more strictly reflect a loss of Cl- secretion. However, this does not discount ENaC as a potentially effective therapeutic target [17].
CF airway disease is manifested through decreased ASL volume, increased mucus viscosity, and impaired mucus elimination by ciliated cells, thereby crippling a primary mechanism of defence in the airways and creating a more static environment for microbial growth. Both infectious [18] and noninfectious stimuli, including interleukin (IL)-1β, IL-17 [19] and neutrophil elastase (NE) [20], drive mucin 5AC (MUC5AC) synthesis [21] that could contribute to mucus accumulation. MUC5AC is important in preventing bacterial adherence in the lung [22] and hypersecretion of MUC5AC alone does not seem to cause respiratory disease [23]. However, in disease states like CF mucin structure, expansion and function may be affected by altered pH [24, 25]. DNA, which is abundant in CF sputum [26], further contributes to mucus viscosity, but may be treated with some success using recombinant DNase I [27, 28].
The sputum of CF patients itself is also conducive to microbial growth as it is quite nutrient rich. Amino acids are found in increased amounts in the sputum of CF patients when compared with healthy controls [29] and correlate with disease severity [30]. Iron, which is sequestered by transferrin, lactoferrin and ferritin, can be found in high amounts in sputum [31, 32], bronchoalveolar lavage fluid (BALF) and lung tissue of the lower airways in CF [33]. Here it can be exploited as an essential cofactor for bacterial enzymes and may also promote biofilm formation [34]. Sputum iron levels were found to correlate positively with the inflammatory cytokines, IL-1β and tumour necrosis factor (TNF)-α and ferritin, but negatively with lung function as measured by forced expiratory volume in 1 s (FEV1). Increases were detected in both stable and acute disease in a sample population that was largely sputum culture positive for Pseudomonas aeruginosa [31]. This increase in iron was observed even in patients who were culture negative for P. aeruginosa, indicating that CF lungs may be more susceptible to infection. In P. aeruginosa positive patients, iron levels correlated positively with bacterial load [32]. Ultimately, these nutrient and iron rich conditions are conducive to microbial growth and may support chronic infection [32, 35]. Mucus in regions of the CF airways has been found to be hypoxic or anaerobic, likely due to increased local aerobic respiration by colonising microbes [9, 36] but also by increased generation of reactive oxygen species by neutrophils [37]. Some microorganisms, such as P. aeruginosa, are capable of efficient growth in anaerobic conditions, but also grow under oxygen-limited microaerobic conditions and anaerobic conditions where denitrification is employed to reduce available nitrate, nitrite and nitrous oxide molecules to nitrogen. Microaerobic or anaerobic growth allows P. aeruginosa to access an additional environmental niche and may also confer the benefit of increased resistance to antibiotics [38] through manipulation of MexEF-OprN efflux pump expression [39].
Secreted host defence molecules augment mucus clearance as a key barrier to microbial survival within the lung. These include a variety of antimicrobials in the ASL, as well as the recruitment of cellular effectors to the site of infection. However, changes to CF airway homeostasis impair these defences, which may be attributed to alterations in pH, protein oxidation, and protease activity. This is illustrated in the pig CF model in which pigs lacking CFTR or expressing ΔF508-CFTR developed lung disease similar to CF in humans [40] and were diminished in their ability to eliminate bacteria [41]. Bacterial killing is reduced in the ASL of CF pigs when compared with controls despite both groups possessing similar concentrations of antimicrobials [42]. This defect in bacterial killing is attributed to a lowered ASL pH, resulting in impaired function of antimicrobials including lactoferrin and lysozyme [43]. β-defensin dysfunction has also been attributed to inactivation by abnormal ASL composition. Although initially attributed to inhibition by high salt concentrations in CF ASL [44], differences in killing between CF and non-CF ASL remained even after desalting procedures, but were rescued by adenoviral expression of CFTR [45]. The importance of pH dysregulation in disease is emphasised by another study in which bicarbonate secretion through the CFTR was required for proper mucin secretion and normalisation of mucus with CF-like properties [46]. Other innate defence mechanisms against microbial growth, including the generation of reactive oxygen and nitrogen species, also appear lacking in CF. Specifically, loss of CFTR in the airway epithelium hinders CFTR-dependent secretion of thiocyanate and its subsequent oxidation to hypothiocyanite, preventing efficient bacterial killing [47]. Thiocyanate levels were found to correlate with better lung function in CF patients, although levels were only slightly reduced in CF pigs and not different in CF patients and controls [48]. Thiocyanate can also serve as an antioxidant, protecting against hypochlorite generation and excessive inflammation [49]. Reduced glutathione (GSH), one of the primary antioxidants in the lung, also undergoes transport into the airways by CFTR [3] and is consequently found to be decreased in BALF from CF airways [50], but not sputum [51]. However, treatment with inhaled glutathione did not significantly improve lung function or decrease exacerbation frequency over 6 months [52]. This was in contrast to an earlier study that found significant improvement in FEV1 with treatment over 2 weeks [53], with decreases in prostaglandin E2 and increases in CD4+ and CD8+ lymphocytes [54]. No reduction in oxidative stress was observed in either trial. The inability of both endogenous and supplementary GSH to control oxidative stress in the CF airways may result in excessive protein oxidation. This has been observed in the BALF of CF patients where increased levels of oxidised proteins were found to correlate with increased airway neutrophil counts and lower lung function [55], although neutrophil/myeloperoxidase-independent sources are also likely to contribute. Oxidative damage may ultimately result in the loss of structure and function in proteins important in host defence, such as surfactant proteins A and D (SP-A and D) [55–57]. Inactivation of antimicrobials may also occur via a protease–antiprotease imbalance in the CF lung [58], in which NE and other proteases have been shown to cleave a variety of targets, ranging from immunoglobulins to complement, CXCR1 and SP-A and D [59]. Nitric oxide, another potent antimicrobial, also appears lacking in CF. This may be due to decreased inducible nitric oxide synthase expression [60, 61], as well as increased expression of the nitric oxide synthase inhibitor asymmetric dimethylarginine [62]. Low levels of nitric oxide may also be attributed to infection by P. aeruginosa and nitric oxide reductase expression [63], which it may utilise to feed into its cycle of anaerobic respiration.
The CF lung environment also affects cellular components of the immune system. Neutrophils, one of the initial cellular responders to infection, have impeded motility due to the thickened CF airway mucus [64] and pathogens such as Staphylococcus aureus and P. aeruginosa may effectively resist phagocytosis through polysaccharide capsule or biofilm formation. This may be further affected by NE, which can directly damage the airways and inhibit the capacity of neutrophils to eliminate bacteria [65]. Loss of CFTR has also been implicated in directly causing cellular abnormalities. For example, CFTR is thought to play a role in the suppression of inflammatory responses via nuclear factor (NF)-κB. This feeds into a hyperinflammatory phenotype, as measured by mitogen-activated protein kinase and NF-κB activation, as well as inflammatory cytokine production in airway epithelial cells and macrophages [66–71]. Increased inflammation was noted even in the absence of observable infection [72, 73]. Neutrophils, a primary cell type in controlling acute infection, are found to accumulate in great numbers in the CF lung. However, they are ultimately incapable of successfully phagocytosing and clearing infection within the CF airway due to both intrinsic defects as well as protease-mediated damage upon entrance into the airways [65]. Neutrophil activation may result in protease, as well as oxidative-mediated damage that may actually worsen airway disease by inactivating secreted antimicrobials and destroying airway structure [74, 75]. These abnormalities are further extended to immune cells, which seem to favour a T-helper cell (Th)2/Th17 phenotype via production of the cytokines IL-1β, 4, 5, 13 and 17 as opposed to a more protective Th1 phenotype against P. aeruginosa [76–78]. It should be noted that it is possible that this skewing is at least due in part to the altered inflammatory milieu created by innate immune dysfunction in CF. For a more comprehensive examination of how the loss of CFTR affects immune function refer to Hartl et al. [75].
It has also been suggested that the CFTR may directly interact with pathogens, serving as a receptor for intestinal epithelial uptake of Salmonella typhi [79] and airway epithelial uptake of P. aeruginosa [80, 81] and Aspergillus fumigatus [82]. Inhibition of binding either through the expression of ΔF508-CFTR or by pretreatment of the microorganism with purified CFTR peptide leads to increased bacterial survival, altered inflammatory cytokine production and cell death. Interactions may also occur without direct interaction with CFTR as in the case of phagosomal escape by Listeria monocytogenes. This process has been shown to be dependent on CFTR channel activity with infection of ΔF508-CFTR expressing macrophages or chemical inhibition of CFTR resulting in phagosomal retention and increased killing of bacteria [83]. Therefore, there is evidence that supports both the localisation and function of CFTR as playing a role in antimicrobial activity. However, the clinical consequences of CFTR localisation versus function have not been clearly correlated with the effects of CFTR genotype on lung disease. This is due to interindividual variability, effects of modifier genes and environmental considerations, which lead to significant variation in lung function even amongst patients with the same CFTR genotype. As summarised in table 1, favourable growth conditions in combination with an ineffective host immune response create a rich, but competitive, environment for a variety of microorganisms in the CF lung.
Infection in CF
Bacterial pathogens
CF airways appear normal at birth but soon become infected by a characteristic group of opportunistic pathogens [42] that includes Haemophilus influenzae, S. aureus, P. aeruginosa, Stenotrophomonas maltophilia and Achromobacter xylosoxidans. This characteristic infection profile tends to follow a distinct trend with age: H. influenzae and S. aureus appear early in life (infants to children), whereas P. aeruginosa infection occurs later in life (adolescence to adulthood). Once acquired, infections can become chronic and last for years. Some CF pathogens have also been observed to be transmitted between individuals, including the P. aeruginosa Liverpool and Australian Epidemic Strains (LES and AES, respectively) [54], Burkholderia cepacia complex [84] and, most recently, Mycobacterium abscessus [85]. The acquisition of P. aeruginosa LES and AES by patient-to-patient transmission was associated with clinical deterioration as measured by lung function and body mass index (BMI) [86]. Co-infection with several of the primary CF pathogens may also modify the course of a chronic CF lung infection, whereby their cohabitation in the CF lung occurs in both mutually beneficial and detrimental capacities. As such, the prevalence of P. aeruginosa appears to correlate inversely with the incidence of S. aureus infection in the CF population [87] and may reflect a mostly antagonistic relationship between the two microorganisms. P. aeruginosa is known to secrete factors that can inhibit the growth of competing S. aureus [88] or to use it as an iron source [89]. Conversely, S. aureus has also been observed to take advantage of growth in proximity to P. aeruginosa whereby the secretion of 4-hydroxy-2-heptylquinoline-N-oxide helps protect S. aureus against aminoglycoside antibiotics and helps select for small colony variants [90].
During the chronic colonisation of CF lungs, P. aeruginosa clones undergo phenotypic conversion. The P. aeruginosa population diversifies in morphotype, motility, virulence, antimicrobial susceptibility and production of adhesins, exopolysaccharides and secondary metabolites [91, 92]. Strains modulate the lipid A structure of lipopolysaccharide (LPS), thereby affecting resistance to host cationic antimicrobial peptides and induction of CXCL8 [93–96]. In addition to sensing LPS through Toll-like receptor (TLR)4, host recognition of P. aeruginosa also occurs by recognition of flagellin through TLR5 [97] and has been shown to be important for bacterial killing by alveolar macrophages [98]. Loss of motility/flagella is common in chronically infecting strains [99]. Modulation of flagellin expression can occur in response to components of CF mucus [100], such as NE [101], allowing for evasion of host recognition and phagocytosis [98, 99]. Flagellin induces the accumulation of myeloid-derived suppressor cells (MDSCs), which have been found to accumulate in CF patients primarily infected with P. aeruginosa. MDSCs can contribute to the evasion of adaptive immunity by suppressing T-cell responses [102]. Chronically infecting strains may also become deficient in quorum sensing and acquire a persister phenotype [103–105]. High mutation rates were observed in lineages that were defective in mismatch repair [106, 107]; however, if hypermutable strains did not emerge in the patients’ lungs, mutation rates remained low [108]. Furthermore, novel adaptive or pathogenic traits can be generated by few or even single mutations provided that predisposing mutational events had occurred before in the clonal lineage [109]. Insight into the microevolution of P. aeruginosa in CF lungs has mainly been gained by whole-genome sequencing of serial isolates [105, 110, 111]. Phenotypic conversion is also often observed in chronic S. aureus infections. Typical traits are hypermutation via insertional inactivation of mutS, biofilm formation and, most frequently, the emergence of small colony variants, which possess higher survival during stationary and post-stationary phases, as well as in the presence of P. aeruginosa and/or antimicrobials [112].
Although lung disease in CF was originally only attributed to specific microbes (S. aureus, and subsequently, P. aeruginosa), it has since been established that standard culture methods do not adequately describe the diversity of microbial populations present in the CF lung [113]. Previously overlooked pathogens are increasingly being identified as significant contributors to CF lung disease including viruses, anaerobic bacteria and fungal pathogens [114]. To aid in this, microbial identification by culture methodology has been supplemented with molecular approaches such as sequencing of ribosomal RNA[113, 115] and restriction fragment length polymorphism analysis [116] in order to obtain a more global view of the CF infection status. Despite providing a more complete snapshot of airway microbial diversity, these methods are also susceptible to false representation by the sample collection method employed. Samples collected directly from lung tissue provide accurate and consistent results, whereas airway washings or expectorated sputum may be prone to contamination by oropharyngeal populations [117]. For clinical purposes, it may be very difficult to avoid contamination as minimisation of invasiveness takes precedence. However, employment of bronchoalveolar lavage (BAL), when possible, could improve accuracy. Studies using molecular approaches of microbial identification have found a statistical correlation with age and infection by H. influenzae, S. maltophilia and P. aeruginosa [118], and relative stability of the CF microbiome, with greater inter- rather than intra-individual variability. Increased disease severity has been positively correlated with reduction in CF microbiome diversity [119–121], which has been attributed to long-term antibiotic use, rather than patient age or lung function [120]. There is now ample evidence that a number of viral, bacterial and fungal pathogens, beyond the dominant P. aeruginosa and S. aureus, can infect and adapt to the CF lung and contribute to disease progression [87, 114].
Viral pathogens
Viral respiratory tract infections in healthy individuals are frequent and often resolve without significant issue. However, they can lead to severe complications in patients, especially children, with chronic respiratory diseases such as CF. Amongst respiratory viruses, rhinoviruses are the predominant viral pathogens associated with pulmonary aggravations in CF, as they are detected in 20–60% of all virus-associated CF exacerbations [122–124]. Elevated levels of inflammatory mediators and the subsequent increases in inflammatory cells have been proposed to explain the increased morbidity caused by respiratory viruses in CF [125, 126]. However, primary airway epithelial cells from healthy and CF patients, infected with rhinoviruses, did not display an increased inflammatory response. There was instead a tendency to lower cytokine levels associated with elevated cell death. This suggests that an elevated inflammatory response upon infection does not explain the increased respiratory morbidity found in CF [127]. Moreover, it has been reported that primary CF airway epithelial cells have an increased susceptibility to respiratory viruses [126, 128]. These findings were confirmed in BAL samples of CF children where rhinovirus load was elevated compared with control patients [129]. Besides their direct pneumopathological effects, respiratory viruses also lead to an elevated susceptibility to bacterial infections through increased bacterial adhesion (reviewed in [130]). For instance, respiratory syncytial virus has been reported to augment the adherence of P. aeruginosa to CF airway epithelial cells [131] and there is evidence that rhinovirus increases H. influenzae, S. aureus and S. pneumonia adhesion to primary healthy nasal epithelial cells [132].
Fungal pathogens
Fungal pathogens now have an increasingly appreciated role in CF lung disease. The most common ones are Aspergillus and Candida species with A. fumigatus and C. albicans as the most prevalent filamentous and yeast fungal species, respectively. Beyond these two prominent species, more exotic genera can also be found in CF airway secretions, such as Penicillium, Alternaria or Scedosporium. Beyond CF, our understanding of the complex age, ethnicity and medication dependent colonisation of the lung with fungi is an area that is starting to evolve. Studies from this field provide the first evidence that even healthy airways are not sterile and contain distinct fungi, termed the “pulmonary mycobiome”. The kinetics, dynamics and disease relevance of the pulmonary mycobiome, however, are poorly understood [133–135].
Despite the increasing detection rate of fungi in CF airways, their pathogenic relevance in the longitudinal course of CF lung disease remains poorly understood and remains, therefore, an ongoing matter of debate. The contribution of A. fumigatus to the allergic disease entity termed allergic bronchopulmonary aspergillosis (ABPA) [136], one form of allergic bronchopulmonary mycosis [137], is appreciated. However, unresolved and controversial questions in the field include: 1) the disease relevance of chronic colonisation/infection with A. fumigatus without the clinical phenotype of ABPA [138], and 2) the relevance of chronic infection with C. albicans [139]. While environmental exposure to fungi is common to CF and non-CF individuals, the impaired mucociliary clearance in CF lungs favours chronic fungal colonisation. As a consequence, the pulmonary immune system is in long-term and intimate contact with fungal allergens that are exposed by the fungus after spore swelling and germination [140]. This host–pathogen interaction is the immunological prerequisite for the establishment of an allergic reaction. Despite the correlation between exposure to Aspergillus spores and the development of ABPA, several studies provide strong evidence that ABPA is primarily the result of a dysregulated host immune response to Aspergillus antigens [76, 141, 142]. The mechanisms by which CF patients become sensitised to the airway microenvironment remain largely elusive, but previous concepts allow speculation that dendritic cells and Th2-associated chemokines and cytokines, such as CCL17/thymus and activation-regulated chemokine and thymic stromal lymphopoeitin, in CF airways play a critical role in this process [142, 143]. Fungi in the airways are mainly sensed by alveolar macrophages, epithelial cells and neutrophils through two distinct pattern recognition receptor (PRR) families: C-type lectin receptors (CLRs), with Dectin-1 as the prototypic fungal CLR PRR, and TLRs [144], with TLR2, TLR4 and TLR6 playing the major roles [145–148]. Engagement of Dectin-1 by whole fungi through the “phagocytic synapse” [149] or fungal components initiates antifungal effector activities, such as phagocytosis [150]. Besides cellular surface receptors, fungi are recognised by humoral PRR, such as SP-D or pentraxin-3, which can be degraded and rendered dysfunctional in the CF airway microenvironment [151, 152].
Regarding adaptive immunity, the Th17 response is essentially involved in anti-fungal host defense [153], with Th17-released IL-17A protein binding fungal cells and causing nutrient starvation [154]. However, over-activation of Th17 responses drives hyperinflammation [155]. This has potential therapeutic implications, as indoleamine 2,3-dioxygenase, a key enzyme of tryptophan metabolism, is involved in Th17 hyperinflammation in CF [78] and tryptophan catabolites (kynurenines) could be therapeutically useful to regulate and dampen hyperinflammation [156, 157]. Vitamin D is an important regulator of innate immunity. The vitamin D pathway has been proposed to be involved in the Th2-driven immunopathogenesis of ABPA in CF patients, where vitamin D deficiency has been identified as an important risk factor. Indeed, it was found that CD4+ T-cells and dendritic cells pulsed with Aspergillus extract and subsequently treated with vitamin D produced less IL-5 and IL-13 [142]. Conversely, Aspergillus derived gliotoxin was shown to impair vitamin D receptor expression and induce the Th2 cytokines IL-5 and IL-13 in bronchial epithelial cells. The effects of gliotoxin were dominant over the effects of vitamin D supplementation on IL-5 and IL-13 secretion. Therefore, supplementation with vitamin D for disease alleviation may require concurrent fungal eradication [158]. Overall, many factors have been found to contribute to the permissive environment of the CF lungs, allowing for infection and persistence by a variety of microorganisms (fig. 1).

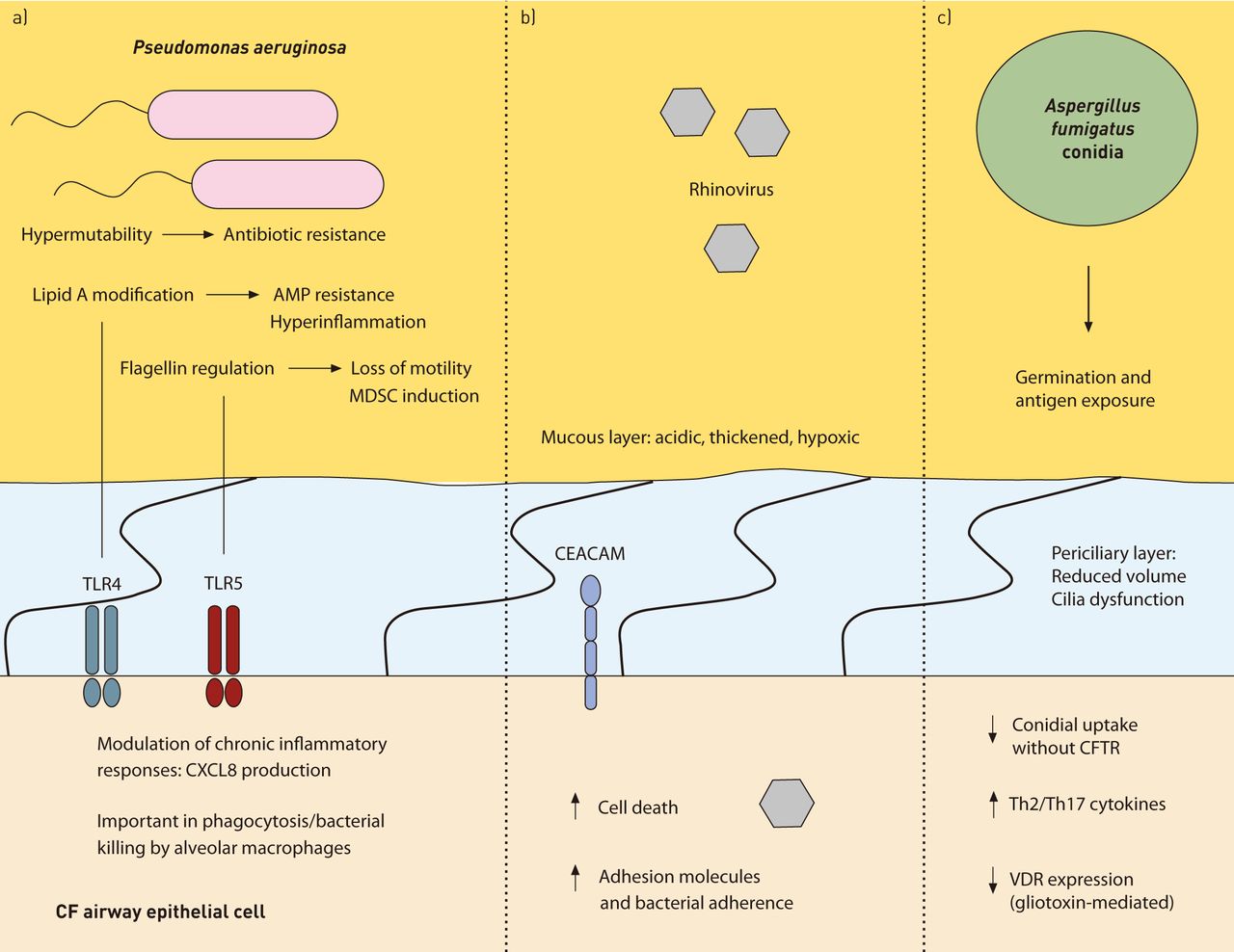{kind=link}
Host–pathogen interactions in cystic fibrosis (CF). This figure illustrates a few of the various interactions between bacterial, viral and fungal pathogens and the CF host. a) Chronically infecting strains of Pseudomonas aeruginosa frequently acquire hypermutable phenotypes through mismatch repair deficiency, encouraging antibiotic resistance and adapted virulence. Inflammatory responses are modulated through changes in lipid A structure and flagellin expression. This provides resistance to host antimicrobial peptides (AMP) and alters CXCL8 production through Toll-like receptor (TLR)4. Loss of motility/changes in flagellin expression can occur in order to evade recognition by TLR5 and phagocytosis by alveolar macrophages. The presence of flagellin can also induce development of myeloid-derived suppressor cells (MDSCs) leading to suppression of T-cell responses. b) Rhinoviral infection may increase expression of cellular adhesion molecules, such as carcinoembryonic antigen-related cell adhesion molecules (CEACAMs), and enhance attachment of bacterial pathogens to host cells. c) Decreased uptake of Aspergillus fumigatus conidia by cystic fibrosis transmembrane conductance regulator (CFTR)-deficient epithelial cells results in reduced clearance and increased cell death. Germination from spore to hyphal form results in increased inflammatory responses. Bronchial epithelial cells induce T-cell-mediated T-helper cell (Th)2 and Th17 responses complicit in allergic bronchopulmonary aspergillosis. This may be further enhanced by A. fumigatus mediated downregulation of the vitamin D receptor (VDR) and increased interleukin (IL)-5/IL-13 production by gliotoxin.
The inflammatory consequences of dysfunctional immune mechanisms in CF: the example of modifier genes
Genome wide association and linkage studies have allowed the identification of genes that appear to play a significant role in modulating disease severity in CF on a population scale (i.e. modifier genes). Interestingly, many of the genes identified have been shown to play direct antimicrobial roles or are important in the regulation of immune responses. This could emphasise the critical role of inflammatory interactions between host and pathogen in worsening lung disease. One of the first identified, MBL2 (mannose-binding lectin (protein C) 2, soluble), recognises N-acetylglucosamine on bacterial cell walls and activates complement for both the direct lysis and recruitment of effector phagocytes to the site of infection. MBL2-deficiency is quite prevalent in the human population [159, 160] and is suspected to predispose individuals to severe respiratory tract infections. Although association of MBL2 polymorphisms with disease severity has been inconsistent in CF populations, meta-analysis of existing studies has found that patients deficient in MBL2 have been associated with worse lung function, earlier acquisition of P. aeruginosa, and increased incidence of death or need for lung transplantation [161]. Furthermore, disease severity was significantly augmented in individuals with MBL2-deficiency who also produced high amounts of transforming growth factor (TGF)-β [162]. Although not directly involved in sensing or combating microorganisms, TGF-β is secreted upon infection and plays an important role, along with the inflammatory cytokines IL-1β and IL-6, in the development of a Th17 inflammatory phenotype which may be responsible for driving excess inflammatory responses in CF epithelia [163, 164] and for increased permissibility to infection by P. aeruginosa [78]. TGF-β also has direct functions in tissue remodelling and may further contribute to worsening lung disease by mediating progressive fibrotic changes to the lung architecture [78] and has itself been firmly established as a modifier gene in CF [165].
Besides modifier genes, biomarker proteins have been shown to predict the course of CF lung disease. Importantly, proteases are already present in early CF lung disease and have recently been demonstrated to predict the course of CF bronchiectasis [166]. However, clinical trials with inhalational α1-antitrypsin did not show efficacy in improving lung function, but did decrease neutrophilic inflammation markers [59, 167]. Further studies, are warranted, but may require a more efficient drug delivery system to ensure adequate lung deposition of α1-antitrypsin. A genetic association with better disease outcomes was also observed in patients possessing polymorphisms for interferon-related developmental regulator 1 (IFRD1) [168, 169]. IFRD1 is important in neutrophil differentiation and controls inflammation through generation of reactive oxygen species and NF-κB p65 translocation. IFRD1-deficient neutrophils were found to be less effective at killing microorganisms. IFRD1 is overexpressed in CF blood neutrophils as compared to controls, although CF airway neutrophils tended to have lower IFRD1 expression, which may be due to the high levels of CXCL8 and CXCL2 typically found in the CF airways [169]. Associations were also detected between the inflammatory genes IL-1β, TLR9, TNF-α, CD95, STAT3 and TNFR in a CF twin/sibling cohort with extreme disease phenotypes. These genes were analysed for their ability to impact Cl- transport across nasal and intestinal epithelia, providing evidence that inflammatory genes do indeed modify the basic defect in CF [170]. Another gene found to modify the basic defect in CF was TLR5, an extracellular sensor for flagellin, which corroborated findings showing reduced TLR5 expression and responsiveness was associated with an improved zBMI score in adults but not children with CF [171].
In addition to candidate gene studies, exome sequencing has been successfully applied to identify a genetic modifier of the age-dependent risk of becoming colonised with P. aeruginosa in CF. The presence of a missense variant in DCTN4 (encodes dynactin-4 protein) was significantly associated with an early age of onset for first and chronic infection with P. aeruginosa, earlier age of onset for first mucoid P. aeruginosa culture and decreased time to conversion from P. aeruginosa positive culture to mucoid P. aeruginosa positive culture [172]. These findings were determined by exome sequencing of individuals with extreme phenotypes of either very early or very late onset of P. aeruginosa infection and subsequent genotyping of an independent non-stratified replication cohort.
Infections in CF and non-CF lung disease
Many other non-CF pulmonary diseases also exhibit chronic opportunistic polymicrobial infection, including diffuse pan bronchiolitis, primary ciliary dyskinesia and chronic obstructive pulmonary disease (COPD). This indicates that infection may be a result of broad dysfunction of pulmonary antimicrobial defences in these diseases. These diseases also possess similarity in the profile of dominant infecting microorganisms (H. influenzae, P. aeruginosa and S. aureus), which may be attributed to the ubiquity and adaptability of these bacteria to persist within the lung microenvironment. Importantly, the nature and patterns of infection do differ between diseases and may reflect subtleties in defective host defence mechanisms or airway conditions. For example, although patients with COPD can be colonised by P. aeruginosa, infections are usually not chronic and are easily treatable [173]. Analysis of the lung microbiome in COPD has also indicated that its composition does indeed change with severity although its makeup is different to that in CF [174]. Inflammatory biomarkers in sputum in general show similarities between CF and COPD and negatively correlate with lung function in both [175]. Additionally, both CF and COPD are characterised by increased numbers of airway neutrophils, antiprotease–protease imbalance and oxidative stress [173], and are modified in disease severity by TGF-β [176, 177]. Despite differences in disease origin and infecting pathogens, this may indicate the involvement of similar pathological processes in these conditions. Further mechanistic definition of which specific pathogens are restricted in certain airway diseases, but not others, may help shed light on the aspects of host defence that confer effective resistance against specific pathogens.
Conclusions and future directions
Having summarised and discussed the complex relationship between host and pathogens in CF lung disease, the following questions remain open to be addressed in the future. How can the CF host response be specifically targeted in order to dampen excessive inflammation without impairing protective immune responses? How do CF airway pathogens regulate each other? Should antibiotic strategies aim to render CF lungs sterile or are certain microbes protective? And if so, is this protective effect age-, CFTR genotype-, gene modifier- and/or medication-dependent? What is the pathogenic contribution of fungi and viruses in the longitudinal course of CF lung disease? Which modifier genes could be helpful in subclassifying CF patients and predicting the response to bacterial infections and disease outcomes?
The combined power of cellular studies, murine models, the CF pig and, most importantly, studies with CF patients will be required to address these complex issues. These different approaches have their individual pitfalls and strengths: murine models of CF do not produce CF lung disease similar to that in humans [178] and substantial differences exist between the murine and human lung architecture. By contrast, pig and ferret models develop lung disease that is more similar to humans [178], but are more expensive. Consequently, translational and multi-methodological research consortiums should pave the way in this research field, with the ultimate goal of improving the length and quality of life of people living with CF.
Footnotes
Support statement: This work was supported by funding from Cystic Fibrosis Canada. S.E. Turvey holds the Aubrey J. Tingle Professorship in Pediatric Immunology and is a clinical scholar of the Michael Smith Foundation for Health Research.
Conflict of interest: Disclosures can be found alongside the online version of this article at err.ersjournals.com
Provenance: Submitted article, peer reviewed.
- Received August 29, 2013.
- Accepted October 7, 2013.
- ©ERS 2014
ERR articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References









