





Abstract
Airway remodelling is a critical feature of chronic bronchial diseases, characterised by aberrant repair of the epithelium and accumulation of fibroblasts, which contribute to extracellular matrix (ECM) deposition resulting in fixed bronchial obstruction. Recently, epithelial–mesenchymal transition (EMT) has been identified as a new source of fibroblasts that could contribute to the remodelling of the airways. This phenomenon consists of the loss of the epithelial phenotype by bronchial epithelial cells and the acquisition of a mesenchymal phenotype. These cells are then able to migrate and secrete ECM molecules. Herein, we review the different types of EMT. We will then focus on the signalling pathways that are involved, such as transforming growth factor-β and Wnt, as well as the more recently described Sonic Hedgehog pathway. Finally, we will highlight the implication of EMT in airway remodelling in specific chronic bronchial pathologies, such as asthma, chronic obstructive pulmonary disease and bronchiolitis obliterans following lung transplantation. Despite the limitations of in vitro models, future studies of EMT in vivo are warranted to shed new light on the pathomechanisms of bronchial obstruction.
Introduction
The epithelial–mesenchymal transition (EMT) refers to a phenomenon in which a polarised epithelium with cell–cell contacts that is attached to the basal membrane differentiates into fibroblast-type mesenchymal cells [1]. This process demonstrates the plasticity of the cells that can also undergo the reverse process, the mesenchymal–epithelial transition (MET) [2]. On the one hand, EMT involves the loss of epithelial polarity due to the disassembly of cell–cell contacts such as adherent junctions (E-cadherin) or tight junctions (zonula occludens-1) and, on the other hand, involves the expression of mesenchymal proteins such as α-smooth muscle actin (SMA), vimentin and/or fibronectin [3, 4]. Alterations in the expression of extracellular matrix (ECM) components (fibronectin and collagens) or degrading enzymes (matrix metalloproteinases (MMP)2 and MMP9) leads to the loss of adherence to the ECM and gives the cells a migratory capacity.
EMT is a physiological phenomenon involved in embryogenesis during the trans-differentiation of epithelial cells into mesenchymal cells [2]. EMT can contribute to the development of cancer, and is implicated in the invasion of tumoural cells and in metastatic migration, as has been demonstrated in models of ovarian, colon, oesophageal and bronchial cancers [5–9].
EMT also participates in healing and tissue repair. The implication of EMT in these processes has been demonstrated in the kidney, the eye and liver fibrosis [10–12]. In inflammatory contexts and during chronic aggression of the epithelium, EMT could generate the fibroblasts needed for the tissue regeneration. However, EMT can persist beyond the inflammation process leading to pathological fibrosis, as in idiopathic pulmonary fibrosis (IPF) [13–15]. In this disease, alveolar epithelial cells are elongated with a modification in the expression of cytokeratins. In addition, these cells are able to secrete pro-fibrotic factors, such as transforming growth factor (TGF)-β, connective tissue growth factor (CTGF), platelet-derived growth factor (PDGF), endothelin-1 and tumour necrosis factor (TNF)-α, and respond to these factors by modifying their morphology and differentiating into fibroblasts [16–22]. Studies in vitro in alveolar cell lines from rats (RLE-6TN), mice (AT2) and humans (A549) have demonstrated the occurrence of EMT in these cells in response to TGF-β [15, 23, 24]. Likewise, alveolar cells can secrete MMPs, contributing to tissue remodelling [25, 26].
Recently, the bronchial epithelium has been studied as a potential source of fibroblasts and myofibroblasts in the chronic remodelling of airways. This bronchial remodelling is one of the main characteristics of diseases such as asthma, chronic obstructive pulmonary disease (COPD) and bronchiolitis obliterans (BO). In these diseases, repeated aggression of the respiratory epithelium (allergens, allogenicity, cycles of infections, cigarette smoke and atmospheric pollutants) leads to chronic inflammation. In response to chronic inflammation, aberrant or uncontrolled tissue repair gives rise to an excessive fibroblastic response contributing to the production of ECM, tissue remodelling and fixed bronchial obstruction. The origin of the fibroblasts responsible for the accumulation of ECM has not yet been determined. They could derive from the proliferation of resident fibroblasts or from the recruitment of progenitor cells.
The role of EMT in these pathologies is not well described but this phenomenon could contribute to the pool of mesenchymal cells, allowing the activation and proliferation of fibroblasts and resident myofibroblasts that are responsible for the accumulation of ECM [27–30]. In this review we propose the description of different types of EMT and will highlight the different signalling pathways involved, including the TGF-β, Wnt and Sonic Hedgehog (Shh) pathways. Finally, we review the implication of EMT in the remodelling of severe chronic inflammatory bronchial diseases such as asthma, COPD and BO following lung transplantation.
EMT: classification and evidence
EMT can be classified into three categories with functional differences. Type I EMT is associated with physiological processes involved in tissue and organ formation during embryogenesis [31]. Type I EMT is implicated in the formation of the mesoderm from the epithelium and in the generation of neural crest cells. In this way, the primary mesenchyma is formed from the epiblast (primitive epithelium). This transition is reversible and the primary mesenchyma can undergo a re-transformation into secondary epithelium. This MET is required for finishing the process of cell differentiation and for defining the three-dimensional structure of the organs. These cycles of EMT and MET are involved in the development of the liver, as well as in the formation of the heart or islets of Langerhans [32–35].
Type III EMT refers to the acquisition of a migratory phenotype by malignant epithelial cells and is associated with tumour invasiveness [36, 37]. During metastasis, epithelial cells lose their polarity and detach from the basal membrane. These cells can migrate through the ECM/tissue and reach the blood circulation to find new metastatic sites to target (fig. 1). Different studies have revealed that metastatic cells express mesenchymal markers such as α-SMA, fibroblast specific protein-1 (FSP1; also known as S100A4), vimentin or desmin [38]. These markers are particularly expressed in the cells localised in the invasion front of the tumour and are associated with the detachment of cells from the ECM and the colonisation of new organs [2, 7, 39].

Epithelial–mesenchymal transition type III; an example of alveolar carcinoma.
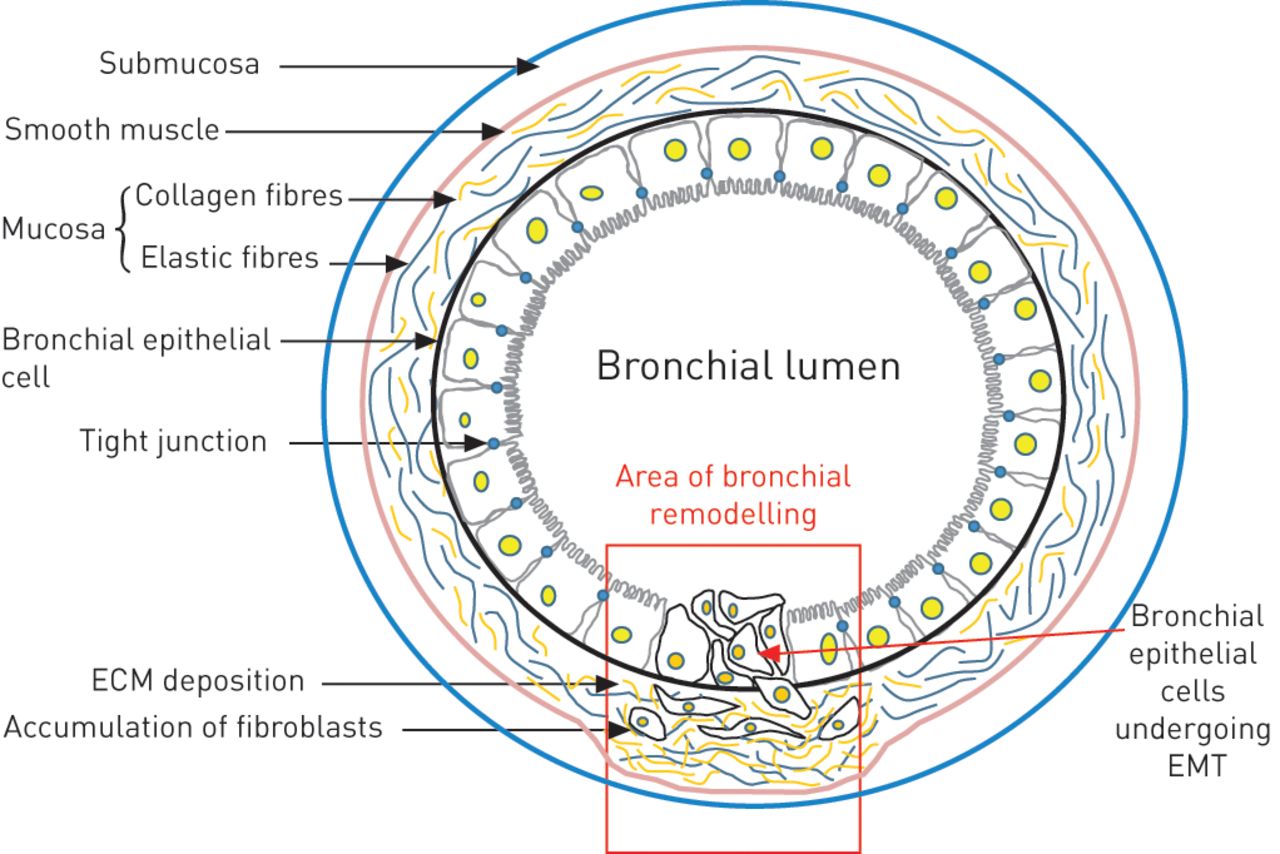
EMT can also participate in the process of healing, contributing to the pool of mesenchymal cells needed for tissue regeneration. However, in response to chronic inflammatory conditions, the processes related to tissue repair become excessive and EMT (type II) is then associated with tissue fibrosis. In this fibrotic process, fibroblasts accumulate secreting collagen fibres, leading to dysfunction of the organ (fig. 2). Using transgenic mice in a model of unilateral ureteral obstruction, Iwano et al. [40] have demonstrated that epithelial cells from the kidney undergo EMT, thus contributing to the fibrosis of the organ. Other studies have shown that EMT is associated with kidney, liver, lung and intestine fibrosis [23, 41].

Epithelial–mesenchymal transition (EMT) type II in bronchial remodelling. ECM: extracellular matrix.
Identification of EMT requires a selection of specific markers. The loss of the epithelial phenotype is characterised by a decrease in the expression of epithelial proteins including junction proteins (E-cadherin and zonula occludens-1), cytokeratins of the cytoskeleton and a decreased expression of the surface protein MUC1 (Mucin 1, cell surface associated). Although the loss of those proteins has been largely described during EMT, the acquisition of mesenchymal markers is more difficult to prove. Markers used for defining the mesenchymal phenotype are vimentin, α-SMA, FSP1, desmin, fibronectin and the production of MMPs [42]. Nonetheless, vimentin is not expressed by all fibroblasts and is also present in leukocytes and endothelial cells [43]. However, α-SMA is expressed by myofibroblasts that represent only a fraction of the activated fibroblasts [40, 44]. Finally, FSP1 (S100A4) is expressed by inflammatory cells, as well as endothelial cells and smooth muscle cells [45–48]. The diversity of mesenchymal markers highlights the complexity of EMT and the need to use different markers to characterise this process.
Induction of EMT and cell signalling
Mechanisms of EMT
The complexity of EMT is due to the diversity of the factors involved and the different effects the factors have depending on the context. The structure of the cytoskeleton can directly induce EMT by disrupting cell–cell or cell–ECM interactions [4, 49] and by inducing the expression of Snail and Slug, regulating the expression of proteins involved in EMT such as E-cadherin and desmoplakin. Mesenchymal markers such as vimentin and fibronectin are upregulated and redistributed [50–53]. The cell environment is very important and pleiotropic signals like reactive oxygen species have an effect in different signalling pathways leading to EMT [54–56]. In addition, EMT can be induced by various growth factors, including fibroblast growth factor (FGF)2, epidermal growth factor, CTGF, insulin-like growth factor (IGF)2, interleukin (IL)-1 and hepatocyte growth factor (HGF). These growth factors have different effects depending on the context in which they are secreted. In the presence of IGF2, β-catenin (proteins linking the cytoskeleton with adherent junctions) dissociates from E-cadherin on the cell surface and is redistributed to the nucleus [57]. FGF2 is considered as a potential factor inducing EMT in kidney cells, and stimulating the secretion of MMP2 and MMP9 which are involved in the degradation of the basal membrane [58]. HGF is one of the growth factors involved in proliferation, migration, differentiation and cell survival. The effect of HGF is associated with the action of the transcription factor Snail, which decreases the expression of E-cadherin and activates mesenchymal genes. HGF is mainly involved in type I EMT, during the formation of somites or cardiac buds [59]. Among the growth factors involved in EMT, TGF-β remains the most studied.
The central role of TGF-β
TGF-β is a cytokine with different roles during development, inflammation, repair, proliferation and cell differentiation. This cytokine has largely been described as an inducer of type I EMT, especially in the formation of neural crests and cardiac valves, and during palatine fusion [60–62]. In pathological conditions such as lung cancer, TGF-β has a dual role. In the first stages of the pathology TGF-β acts as a tumour suppressor, but in the late stages of cancer TGF-β favours type III EMT contributing to the dissemination of metastases. In vitro studies in different pulmonary epithelial cells obtained from healthy donors have shown that exposure to TGF-β induces a mesenchymal phenotype in these cells [29, 63–68].
TGF-β is a ubiquitous cytokine that is present in several isoforms (TGF-β1, TGF-β2, and TGF-β3). TGF-β binding induces the dimerisation of the type I and II TGF-β receptors. The type II receptor, which has constitutive kinase activity, phosphorylates the type I receptor activating the canonic TGF-β signalling cascade involving the intracellular proteins of the Smad family. Phosphorylated TGF-β receptors phosphorylate and activate Smad2 and Smad3 [69]. The Smad2/3 dimer forms a complex with Smad 4, named the Smad protein complex. This complex then translocates to the nucleus where it participates in the transcriptional regulation of target genes [70, 71] (fig. 3). The Smad complex then represses the expression of E-cadherin by the transcription factors Snail1 and Snail2 (snail and slug, respectively) [50, 72]. Snail induces the expression of mesenchymal proteins like N-cadherin, fibronectin, vitronectine and MMPs (fig. 3) [73, 74].

{kind=link}
{kind=link}
{kind=link}
Cell signalling involved in epithelial–mesenchymal transition. ZO: zonula occudens; Wnt: wingless tail; Hh: Hedgehog; SMO: Smoothened; GSK-3β: glycogen synthase kinase-3β; Ptch: Patched; MAPK: mitogen activated protein kinase; PI3K: phosphatidylinositol 3-kinase; Zeb: zinc finger E box binding homeobox; MMP: matrix metalloproteinase; TGF: transforming growth factor; BMP7: bone morphogenetic protein 7; P: phosphate.
Using Smad3 knock-out mice, Zavadil et al. [75] have shown that Smad3 was necessary for EMT induction by TGF-β [75]. Furthermore, recent studies have demonstrated that Smad 3 controls the majority of genes involved in EMT [38]. Thus, inhibition of the TGF-β pathway appears to be an interesting therapeutic strategy for inhibiting EMT. Indeed, inhibitors of TGF-β or its receptors decrease metastasis and/or affect the invasive properties of cells in vivo [76–78]. There are some endogenous proteins that act as inhibitors of TGF-β signalling. For instance, Smad7 is associated with the activated receptor TGF-β1, preventing the phosphorylation of Smad2/3. Bone morphogenetic protein (BMP)7, a member of the TGF-β superfamily, uses TGF-β receptors to induce the formation of a Smad complex (Smad 1 and 4) and its nuclear translocation (fig. 3). This complex represses the transcription of EMT Zeb1 (zinc finger E box binding homeobox) and Snail1 transcription. In a murine model of kidney fibrosis, EMT induced by TGF-β can be inhibited by BMP7 [79–81]. However, in the bronchial epithelial cells (BEC) BMP7 is not effective for reverting EMT [63]. This suggests that EMT mechanisms depend on the cell type and tissue [82]. Recently, Gardner et al. [65] identified a new therapeutic target, TAK1, which could be involved in the induction of EMT in the bronchial epithelium by TGF-β and TNF-α.
The induction of EMT by TGF-β is mainly dependent on Smad but can also implicate other pathways, acting in synergy with Smad signalling. Indeed, TGF-β can activate p38, mitogen activated protein kinase and phosphatidylinositol 3-kinase pathways implicated in type III EMT (fig. 3) [83, 84]. The RhoA pathway can be inhibited by TGF-β, destabilising tight junctions. The mitogen activated protein kinase pathway (Ras/Raf) contributes to the autocrine secretion of TGF-β [85]. In some cases the activation of these proteins is necessary, but not sufficient, for inducing EMT [86].
Other signalling pathways involved in EMT
Wnt ligand
The β-catenin pathway, induced by the ligand Wnt (wingless tail), is involved in the mechanisms of lung remodelling in pathological conditions [87–90]. Briefly, the ligand Wnt binds to the receptor Frizzled (Fzd) inducing its phosphorylation and the inactivation of glycogen synthase kinase-3β. This contributes to the cytosolic accumulation of β-catenin and its translocation to the nucleus (fig. 3), where it can interact with lymphoid-enhancer binding factor and T-cell-specific transcription factor, inducing the transcription of target genes, such as those encoding C-myc and Cyclin D [91]. Thus, leading to a modification of cytokeratins expression and the reorganisation of the cytoskeleton. The canonical Wnt pathway has been described as a mediator of TGF-β signalling in alveolar type II cells and pulmonary fibrosis [92, 93]. Other signalling pathways such as Notch, nuclear factor-κB and Shh have also been shown to participate in EMT [86, 94, 95].
Hedgehog signalling
The Hedgehog gene (Hh) was first identified during a screening of genetic mutations involved in the segmentation of Drosophila larvae [96]. Orthologs of Hh have been described since in the vertebrates. There are three genes coding for the ligand Hh in vertebrates: Shh, Indian Hedgehog (Ihh) and Desert Hedgehog (Dhh). Among these three genes, Shh is the most broadly expressed in mammalian tissues [97]. The presence of Shh releases Smoothened (SMO) from the inhibition exerted by the receptor Patched (Ptch). SMO is a G protein associated with the receptor that transduces Hh signalling inside the cell (fig. 3). The activation of the Shh pathway results in the nuclear translocation of the transcription factor Gli (fig. 3), which regulates the expression of genes involved in proliferation, such as cyclins, and genes of the Hh pathway itself, such as Gli and Ptch [98]. The Shh pathway is activated during lung repair and the physiological development of the lung [99]. During animal development, Shh is expressed in the airway epithelium and is essential for regulating epithelial–mesenchymal crosstalk in the lung. The Hh pathway is essential for lung formation as Shh knockout embryos exhibit severe defects in the development of the lung due to failure of branching and tissue growth [100–102]. Moreover, Gli2-knockout mice die at birth with lungs presenting a reduced size and defects in the ramification [103]. In adult tissues, growing evidence has highlighted the importance of the Hh pathway in cell migration. The canonical Hh pathway, dependent on SMO and the transcription factor Gli, is also implicated in cell migration and invasion through the regulation of genes associated with EMT. The knockout of Gli1 reduces the expression of Snail and MMP9 and increases the expression of E-cadherin [104]. In addition, when the expression of Gli1 is induced in epithelial cells (using a tetracyclin-controlled transcriptional activation system), Snail is expressed in a fast and considerable manner [105]. In mice, the overexpression of Gli1 in the skin induces the formation of skin lesions and the loss of E-cadherin expression [105]. In pancreatic cancer cells, expression of E-cadherin is independent of Snail and Slug but directly regulated by Gli1 [106]. The inhibition of the Hh pathway with cyclopamine, a steroid that blocks SMO, decreases the expression of Gli1 and Gli2, but also the expression of Sip1, Snail2 and Twist2 [107]. Thus, the Hh pathway, through canonical and non-canonical signalling, is involved in the regulation of genes associated with EMT and regulates cell migration. The impact of the Hh pathway in cell migration and mesenchymal transformation can have important consequences in pathological processes such as fibrosis. In mice models of nonalcoholic fatty liver disease, EMT in the liver occurs with the concomitant activation of the Hh pathway and is associated with liver fibrosis. Furthermore, in Ptch+/- mice, the over-activation of the Hh pathway parallels an increase in EMT and an aggravated liver fibrosis [108]. In the lung, expression of the Shh ligand is increased in patients with usual interstitial pneumonia, nonspecific interstitial pneumonia [109], cryptogenic pneumonia [110] or IPF [111, 112]. Recent studies in IPF lungs have revealed distinct expression of Hh-related proteins in IPF tissues [111, 112]. Shh is expressed in bronchial and alveolar epithelial cells in fibrotic areas while Ptch1 is observed in fibroblasts, interstitial inflammatory cells and the hyperplastic epithelium. The G protein-coupled receptor SMO is detected in the hyperplastic epithelium, interstitial inflammatory cells and mesenchymal cells, forming fibroblast foci in IPF tissues. The Hh-specific transcription factors, Gli1–3, are found in IPF lungs with a different pattern of expression. While Gli1 is detected in the nucleus of epithelial cells and fibroblasts and in inflammatory cells, Gli2 has a nuclear distribution in alveolar epithelial cells and Gli3 shows a weak expression in fibroblastic foci. Together, these data show that Hh-related molecules are highly expressed in IPF lungs, indicating an activation of the Hh pathway in this type of lung fibrosis. Moreover, in fibroblasts, Shh increases the synthesis of proteins related to ECM, such as collagen and fibronectin [112, 113]. Interestingly, inhibition of the Hh pathway reduces the expression of α-SMA, collagen 1 and fibronectin and abrogates the effect of TGF-β on these proteins [111], highlighting the importance of the TGF-β/Hh crosstalk that may promote a mesenchymal phenotype in pathological contexts. In different types of lung cancer, the Shh pathway has also been found to be re-activated [99, 113–116], and is involved in the mesenchymal transition of tumoural cells associated with the formation of metastases. In a model of lung metastases, colon carcinoma cells injected into nude mice induced pulmonary lesions. In this model, the expression of a short hairpin (sh)RNA against SMO inhibits the metastatic growth. In addition, cells from colon carcinoma expressing shRNA against Ptch1 or Gli1 lose epithelial morphology, and increase their capacity for migration and expression of EMT-related genes, such as FOXC2, vimentin, Snail1 and ZFHX1B, while the expression of E-cadherin is decreased [117]. Shh can stimulate the small GTPases Rac1 and RhoA, resulting in the migration of the cells in a SMO-dependant but Gli-independent mechanism, which is therefore related to a “non-canonical” Hh pathway [118, 119].
In summary, studies in the lung and other organs have revealed the involvement of the Shh pathway in EMT in vitro and in vivo. The impact of Shh in cell migration can also be due to the interaction between this pathway and other signalling cascades such as TGF-β, one of the main factors involved in EMT. Evidence has shown an interaction between the Shh and TGF-β Smad pathway during development, but also in adult tissues [111, 120–122]. This type of interaction can enhance EMT effects in bronchial chronic lung diseases.
EMT in chronic bronchial diseases
EMT and allergic asthma
Asthma is characterised by the remodelling of airways, including subepithelial fibrosis, hyperplasia of myofibroblasts and myocytes, and an increase in smooth muscle fibres [123]. In histological samples from asthmatics, myofibroblasts have been found in close proximity to smooth muscle and lamina reticularis [124, 125]. However, the origin of these myofibroblasts remains unclear. Recently, the involvement of EMT in the bronchial remodelling of asthmatic patients has been proposed [40].
The respiratory epithelium constitutes a physical barrier protecting the organism against infections, harmful particles or allergens. After chronic aggression, the epithelium is able to modulate the innate immune response and adaptive immune response by interacting with cells from the immune system, but also by secreting soluble factors such as TGF-β, epidermal growth factor, FGF2, IGF1, PDGF and endothelin-1 [126, 127]. All these modifications contribute to the proliferation and differentiation of fibroblasts and myofibroblasts, and to the accumulation of fibronectin and collagen in the lamina reticularis leading to the thickening of the bronchial wall. Studies in different models of allergic asthma in vivo and investigations in vitro with BEC of asthmatic patients have shown that circulating progenitors of fibroblasts (fibrocytes) are recruited during the repair of the epithelium. In bronchial biopsies, Nihlberg et al. [128] have found that fibrocytes present in the basal membrane of patients with moderate asthma correlated with the thickening of this membrane. This type of fibrocyte was also found in the bronchoalveolar lavage (BAL) of these patients suggesting that circulating fibrocytes differentiated into myofibroblasts [128]. Indeed, it is not clear whether the fibrocytes are able to differentiate into myofibroblasts or whether they stimulate the differentiation and proliferation of other progenitors or mesenchymal resident cells.
With the aim of identifying EMT in vivo, Johnson et al. [129] have developed a murine transgenic model in which airway epithelial cells express the lac-Z reporter gene. After 5 days of exposure to house dust mite allergen epithelial cells underwent EMT, co-expressed the protein S100A4 and accumulated in the smooth muscle. Other epithelial cells co-expressing vimentin were found in the subepithelial region. These results suggest that EMT is implicated in the bronchial remodelling associated with asthma [129]. This is in agreement with previous reports demonstrating that exposure of epithelial cells to proteolytically active allergens such as Der p 1, a major mite allergen [64, 130], or to some pollen allergens [131] induced the degradation of the epithelium. Other studies in bronchial cell lines exposed to mite extracts have shown that the proteolytic action of this allergen occurs in the cell–cell contacts, releasing β-catenins from adherent junctions [64]. Compared with TGF-β, mite extract alone is not able to induce EMT. Yet in combination with TGF-β, it enhances the expression of EMT markers. These results highlight the importance of TGF-β in EMT in asthma [64, 129] and elevated levels of TGF-β have been reported in BAL and bronchial biopsies of asthmatic patients [132, 133]. The increase of TGF-β correlates with airflow obstruction and airway wall thickening, and is not changed despite oral treatment with corticosteroids [134, 135]. Finally, Hackett et al. [63] demonstrated the induction of EMT in primary epithelial cells differentiated in the air–liquid interface after TGF-β exposure. In this model of culture, EMT was localised in basal cells (identified by cytokeratin 5 and p63 expression) [63]. Moreover, EMT was dependent on Smad3. The inhibition of Smad3 by RNA interference stabilised E-cadherin expression and avoids expression of mesenchymal proteins such as fibronectin. Nonetheless, the role of TGF-β1 in asthma-associated EMT is not completely understood. For instance, treatment of BEC with BMP7, an inhibitor of TGF-β, does not reverse EMT, as is the case in murine models of kidney fibrosis. This could be due to a dose-dependent effect of BMP7, an organ/tissue specific effect [63, 67], or other signalling pathways involved in asthma. In an in vitro model using BEC and the Beas-2b cell line, Doerner et al. [67] suggested a synergistic effect between TGF-β and IL1-β. Indeed, the culture of bronchial primary cells with IL-1β induced a decrease in the expression of E-cadherin and an increase in the expression of tenascin C, a component of ECM. Co-treatment with TGF-β and IL-1β accentuated these changes. These results suggest that the inflammatory context plays an important role in EMT. Nevertheless, in the same study, glucocorticoids did not have any effect in EMT. Future studies should elucidate the role of inflammatory pathways insensitive to glucocorticoids such as IL-17, TNF-α or interferon-γ in EMT.
EMT and COPD
COPD is characterised by the progressive obstruction of the airways, mainly due to inflammation induced by the inhalation of cigarette smoke [136–138]. Chronic exposure to cigarette smoke induces the infiltration of inflammatory cells in the mucosa, sub-mucosa and glandular tissue. This causes the secretion of mucus by hyperplasic epithelial cells and thickening of the wall of small airways [139]. Remodelling of the small airways and emphysema contribute to progressive deterioration of respiratory function [140]. Mechanisms leading to these lesions are not yet clearly understood [15, 141]. Recently, Sohal et al. [141] suggested the implication of EMT in COPD, showing that the basal membrane was altered more in COPD patients who smoked compared with smokers with normal lung function and non-smoking controls. Cells expressing MMP9 and S100A4 were found in the fragmented areas, revealing proteolytic activity and a fibroblast phenotype. These results suggest that fragmentation of the basal membrane could contribute to EMT in vivo [31, 142–144]. Some immune cells (CD4+ and CD8+ lymphocytes, macrophages and dendritic cells) were found in the lamina reticularis and in the lamina propia. Cells localised in the fragmentation area of the lamina reticularis poorly expressed cytokeratine, an epithelial marker, and S100A4 [141]. In addition, membrane fragmentation correlated with smoking history in COPD and stopping smoking could reduce the migratory capacity of fibroblastic cells [145]. These studies only used immunohistochemical staining from patient biopsies; therefore, the results do not show whether the cells identified as fibroblasts are from the migration of progenitor cells or from the ability of the epithelium to differentiate into fibroblast-like cells. Although these studies are the first in favour of the implication of EMT during tissue remodelling in COPD, complementary studies are needed to follow in vivo the modification of the epithelial phenotype and cell migration. This type of study would allow a positive control for EMT to be included and to determine whether fragmentation of the lamina reticularis constitutes a key step in COPD progression.
EMT and BO following lung transplantation
Lung transplantation is the only therapeutic option for patients with terminal respiratory failure. Despite the improvement in immunosuppressive treatments and surgical techniques, chronic lung dysfunction, also called BO, is the major limitation of lung transplantation. BO is an irreversible condition with a prevalence of ∼50 and ∼75% at 5 years and 10 years post-transplant, respectively [146]. It is considered to be the result of chronic rejection.
BO is characterised by distal inflammation and fibrosis of the alveoli and bronchi, a process that contributes to bronchial obstruction [147]. BO results from an allogeneic mechanism in which allo-antigens are presented to T-lymphocytes from the recipient [148, 149]. Even though the immune response plays a role in the development of BO, the respiratory epithelium also contributes to the fibrotic phenomenon. Some studies have described BEC as the major target of the immune response. These cells play a central role in the development of BO [150–152]. Mauck et al. [150] have shown in vivo that BEC can induce an allo-immune response and express various growth factors that play a role in the development of BO. These results were confirmed by Jaramillo et al. [151], showing that antibodies against human leukocyte antigen class I are able to activate BEC to secrete fibrotic factors.
The epithelial injury leads to epithelium repair, which could contribute to the fibrotic process when excessive or inappropriate [147, 153, 154]. The cellular mechanisms involved during bronchial remodelling in BO are not clearly understood. Lung fibroblasts can be derived from fibroblasts in situ or from the recruitment of circulating precursors in the site of inflammation. Recent studies suggest that EMT is implicated during the development of BO [29, 30, 155, 156], as has been demonstrated for chronic kidney rejection [157–160]. A study with biopsies and ex vivo cultures from lung transplant recipients showed that BEC expressed S100A4 [30]. Secretion of MMP9 was also increased following TGF-β1 treatment and BEC were able to invade a collagen gel. This study is the first to demonstrate the effect of TGF-β in BEC from lung transplant recipients. It would be interesting to undertake a kinetics study of such cultures in order to determine if the modifications observed in the cell phenotype correspond to a process of tissue repair or to a pathological fibrotic process. In this context, this type of investigation could identify predictive markers of lung remodelling associated with BO. Although this study does not prove a direct link between EMT and BO, it underscores the plasticity of BEC from lung transplant patients. Using flow cytometry, Hodge et al. [156] have demonstrated the increased expression of α-SMA and extra domain-fibronectin in BEC from BO patients compared with lung transplant patients without BO. After longitudinal follow-up, the expression of S100A4, α-SMA and extra domain-fibronectin was increased [156]. These results, which are yet to be confirmed with a higher number of patients, encourage continuation with such investigations in order to determine if there is a link between myofibroblastic markers and BO.
Studies in the inflammatory environment of BO have shown that TGF-β is one of the most important factors involved in the fibrotic process and high levels of TGF-β are found in the BAL from BO patients [148, 161–165]. Other pro-inflammatory cytokines, such as IL-8, TNF-α and IL-1β, are also highly expressed in BO patients [127, 148, 165]. This inflammatory environment could contribute to EMT in BO.
Borthwick et al. [29] reported that BEC isolated from BO patients could undergo EMT, characterised by a decrease in the expression of E-cadherin and an increase in the expression of vimentin and α-SMA. Co-treatment with TGF-β and TNF-α accentuates the mesenchymal phenotype. Other inflammatory cytokines such as IL-1β and IL-8 enhance the process of EMT [29]. HGF has also been found in the BAL of BO patients [151]. This growth factor plays a role in the repair of the epithelium following injury and can partially induce EMT [143, 166]. The dual role of HGF makes it difficult to determine whether it is involved in a physiological EMT during tissue repair or in pathological EMT participating in tissue fibrosis. Other factors from the environment associated with lung transplantation can also play a role in EMT of BO patients. Bacterial infection with Pseudomonas aeruginosa has been shown to significantly increase inflammation in lung transplant recipients [167]. Borthwick et al. [168] have shown that bacteria are able to induce EMT in the presence of TGF-β. This effect is indirect and due to the activation of immune cells [168]. It would be interesting to investigate whether infection with cytomegalovirus, which constitutes a risk factor for BO, can induce EMT. In the chronic rejection of the kidney, immunosuppressive treatments have been described to be involved in EMT [169].
These studies with BEC from lung transplant patients do not allow the origin of fibroblasts contributing to the bronchial obstruction to be determined. This highlights the difficulty in identifying EMT in ex vivo models. Other studies in vitro are necessary to test whether other risk factors (e.g. cytomegalovirus infection or the allogenic context) could contribute to EMT. It would also be interesting to investigate the kinetics of BEC cultures and to correlate the profile of the cells with the clinical stage of lung transplant recipients. It would be then possible to determine if different mesenchymal proteins could be considered as predictive markers of remodelling in BO.
Conclusions and outlook
The studies presented here show that EMT is implicated in the bronchial remodelling in chronic pathologies such as asthma, COPD and BO. Given the diversity of markers used to characterise EMT and the variety of study models, the occurrence of EMT is not always easy to demonstrate. Longitudinal studies from patient samples will help determine whether EMT is involved in tissue repair or in a late process contributing to the fibrosis of the tissue.
Studies performed during organ development and metastatic processes have revealed critical signalling pathways involved in EMT. Regarding bronchial pathologies, the same signalling pathways seem to be implicated with a different regulation. It would be of great interest to pursue these studies to determine the mechanism involved in EMT in each bronchial disease and, therefore, distinguish potential therapeutic targets to limit bronchial remodelling.
Footnotes
Support Statement: The research programme is supported by the Institut Thématique Multi Organisme Immuno-Hémato-Pneumologie of the French National Alliance for Life Sciences and Health (Paris, France), Vaincre la Mucoviscidose (Paris), Agence de Biomédecine (Paris), INSERM (Paris), Région Pays de La Loire (Nantes, France), Institut de Recherche en Santé Respiratoire des Pays de la Loire (Nantes) and Fondation du soufflé (Paris).
Provenance: Submitted article, peer reviewed.
Statement of Interest: Disclosures can be found alongside the online version of this article at err.ersjournals.com
- Received June 13, 2013.
- Accepted July 10, 2013.
- ©ERS 2014
ERR articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 3.0.
References









