





Abstract
Pirfenidone is an orally active, small molecule that inhibits synthesis of profibrotic and inflammatory mediators. It was approved for the treatment of adults with mild-to-moderate idiopathic pulmonary fibrosis in the European Union based on the results of two pivotal phase III, double-blind, randomised, placebo-controlled clinical trials (CAPACITY) demonstrating efficacy and safety, and supported by two Japanese clinical trials (SP2 and SP3). Currently, there is increasing interest in experience with pirfenidone in patients relating to the real-world setting.
Following the publication of the CAPACITY clinical studies, additional analyses have been conducted to provide further support for pirfenidone in clinical practice, including a modified per-protocol analysis of the CAPACITY study population. New data from the RECAP extension study also provided longer term data for pirfenidone and promising continuation rates with treatment. Pirfenidone is also being evaluated in specialist centre cohorts providing important information on real-world efficacy and safety. Increasing experience with pirfenidone in everyday clinical practice is helping to establish \expert guidance on the management of known adverse events, together with practical recommendations, to ensure adherence to treatment so that the possible longer term benefits of pirfenidone treatment in reducing lung function decline can be maximised.
Introduction
Idiopathic pulmonary fibrosis (IPF) is the most common type of idiopathic interstitial lung disease and is associated with progressive scarring of the lung interstitium resulting in irreversible loss of lung function [1, 2]. The clinical course of IPF is chronic, progressive and invariably fatal, with a mean survival of 2–5 years following diagnosis [3].
The search for effective treatments for IPF has involved numerous multicentre, randomised, placebo-controlled trials investigating agents with different mechanisms of action. Although many of these clinical trials failed to demonstrate a statistically significant treatment effect, as recently reviewed by Cottin [4, 5] and Antoniou et al. [6], positive results from two pivotal phase III trials (the CAPACITY (Clinical Studies Assessing Pirfenidone in IPF: Research of Efficacy and Safety Outcomes) programme) and two supportive Japanese trials in patients with IPF led to pirfenidone becoming the first agent to be approved for the treatment of patients with mild-to-moderate IPF in the European Union in 2011 [7–9]. Pirfenidone is also approved in Canada, Japan, India, China, South Korea and Argentina. The US Food and Drug Administration requested an additional phase III clinical trial to support regulatory registration in the USA, which is currently ongoing (ASCEND (Assessment of Pirfenidone to Confirm Efficacy and Safety in IPF); www.ClinicalTrials.gov identifier NCT01366209).
The rationale for pirfenidone treatment in IPF was initially based on in vitro and in vivo preclinical studies that demonstrated anti-fibrotic and anti-inflammatory properties [10]. To date, there have been four randomised controlled trials (RCTs) investigating the efficacy and safety of pirfenidone in IPF, including the CAPACITY programme and two studies conducted in Japan (SP2 phase II and SP3 phase III) [7–9].
CAPACITY consisted of two, concurrent, placebo-controlled phase III clinical trials (studies 004 and 006) that evaluated pirfenidone efficacy and safety in a total of 779 patients with mild-to-moderate IPF [7]. The primary end-point in the CAPACITY trials was change in forced vital capacity (FVC) % predicted from baseline to week 72. Pirfenidone significantly reduced mean decline from baseline in FVC % pred compared with placebo at week 72 in study 004 (-8.0% versus -12.4%, respectively; p = 0.001), as well as the proportion of patients with an FVC decline ≥10% [7]. Although study 006 recorded a significant treatment difference in FVC % pred up to week 48, this was not maintained at week 72. The difference between primary end-point results at week 72 for study 004 and study 006 is unclear but may have resulted from a modest attenuation in the rate of FVC decline in the placebo group after week 48 in study 006, although other factors such as genetic differences between groups (e.g. MUC5B promoter polymorphism) or the greater proportion of patients with obstructive airway disease in the respective placebo arm should be considered. Pre-specified pooled analyses of the primary and secondary efficacy outcome measures were also conducted based on a combined analysis of the pirfenidone 2403 mg group compared with the placebo group across both studies. These analyses are considered exploratory because the primary end-point of both studies was not met. Pooled data for the primary end-point showed a significant treatment effect with pirfenidone, with a mean change in FVC % pred from baseline of -8.5% for pirfenidone versus -11.0% for placebo (p = 0.005). In the pooled analysis, a positive treatment effect for pirfenidone versus placebo was also observed in the secondary end-points of progression-free survival (defined as time to confirmed ≥10% decline in FVC % pred, ≥15% decline in diffusing capacity of the lung carbon monoxide % pred, or death), categorical decline in FVC ≥10% and the 6-min walk test (6MWT) distance [7]. Pirfenidone was generally well tolerated, with the most common adverse events being gastrointestinal and skin related [7].
The clinical efficacy of pirfenidone is further supported by a Cochrane meta-analysis of three phase III studies (i.e. the Japanese SP3 trial and the two phase III CAPACITY studies), which demonstrated that treatment with pirfenidone reduced the risk of disease progression or death by 30% (HR 0.70, 95% CI 0.56–0.88; p = 0.002) [11].
Following European approval, pirfenidone has been introduced into clinical practice for the treatment of adult patients with mild-to-moderate IPF and there is increasing interest in the efficacy and tolerability of pirfenidone in the real-world setting. Here, additional analyses and data which may reflect the use of pirfenidone in everyday practice are reviewed, including a per-protocol analysis of the CAPACITY studies, longer term outcomes in the CAPACITY extension study RECAP, and experience with pirfenidone from single-centre cohorts. However, it must be acknowledged that per-protocol analyses may be subject to bias, and extension studies and case series might be hampered by selection and publication bias.
Efficacy of pirfenidone: CAPACITY per-protocol analysis
Pirfenidone efficacy and safety was evaluated in the CAPACITY studies using intention-to-treat (ITT) analysis [7], which is considered the primary population for analysis in superiority trials. However, the ITT analysis, which includes patients’ permanently discontinuing treatment before the study termination, may provide a more conservative estimate of treatment effect.
A recent per-protocol analysis of the CAPACITY studies has been conducted which included all randomised patients stratified into completers and non-completers. Completers were defined as all patients completing 72 weeks of treatment and patients who died during treatment or within 28 days after the last dose. Results from this per-protocol analysis may, to a certain extent, be more reflective of the real-world population, and consistent conclusions from both ITT and per-protocol analyses increase the confidence in treatment outcomes in everyday practice. In the pooled analysis of CAPACITY studies 004 and 006 there were 86.7% (n = 299) completers in the pirfenidone group compared to 91.6% (n = 318) in the placebo arm. The mean baseline FVC % pred (75.0% and 74.6% for completers in the pirfenidone and placebo groups, respectively) and 6MWT distance (396 m and 407 m for completers in the pirfenidone and placebo groups, respectively) were similar to the baseline characteristics in the CAPACITY ITT analysis and are representative of IPF cohorts encountered in clinical practice (table 1) [7].
Evaluation of the primary end-point in CAPACITY studies (FVC decline from baseline to week 72) demonstrated similar and consistent outcomes for both the per-protocol and ITT analyses, with an absolute difference in decline in FVC % pred of 3% in the per-protocol analysis (30% relative reduction) and 2.5% in the ITT analysis (23% relative reduction). As in the original ITT analysis, the mean change in FVC decline with pirfenidone was significantly less than with placebo at each time-point from week 12 to week 72 in this per-protocol analysis (fig. 1). Consistent and sustained effects in the per-protocol analysis were also observed for pirfenidone across secondary end-points. At week 72, the per-protocol analysis demonstrated that 17.4% of patients in the pirfenidone group versus 26.9% in the placebo arm had a ≥10% decline in FVC % pred representing a relative reduction of 30% (fig. 2). This is consistent with the ITT analysis (21% in the pirfenidone group versus 31% in the placebo group; p = 0.003) [7]. A greater relative reduction in decline in 6MWT distance with pirfenidone was seen in the per-protocol analysis, with an absolute decline in 6MWT distance of 27 m (39% relative reduction versus placebo) compared to 24 m in the ITT analysis (31% relative reduction versus placebo). It has previously been reported that change in 6MWT distance is a clinically useful measure of disease status and risk of mortality [12].
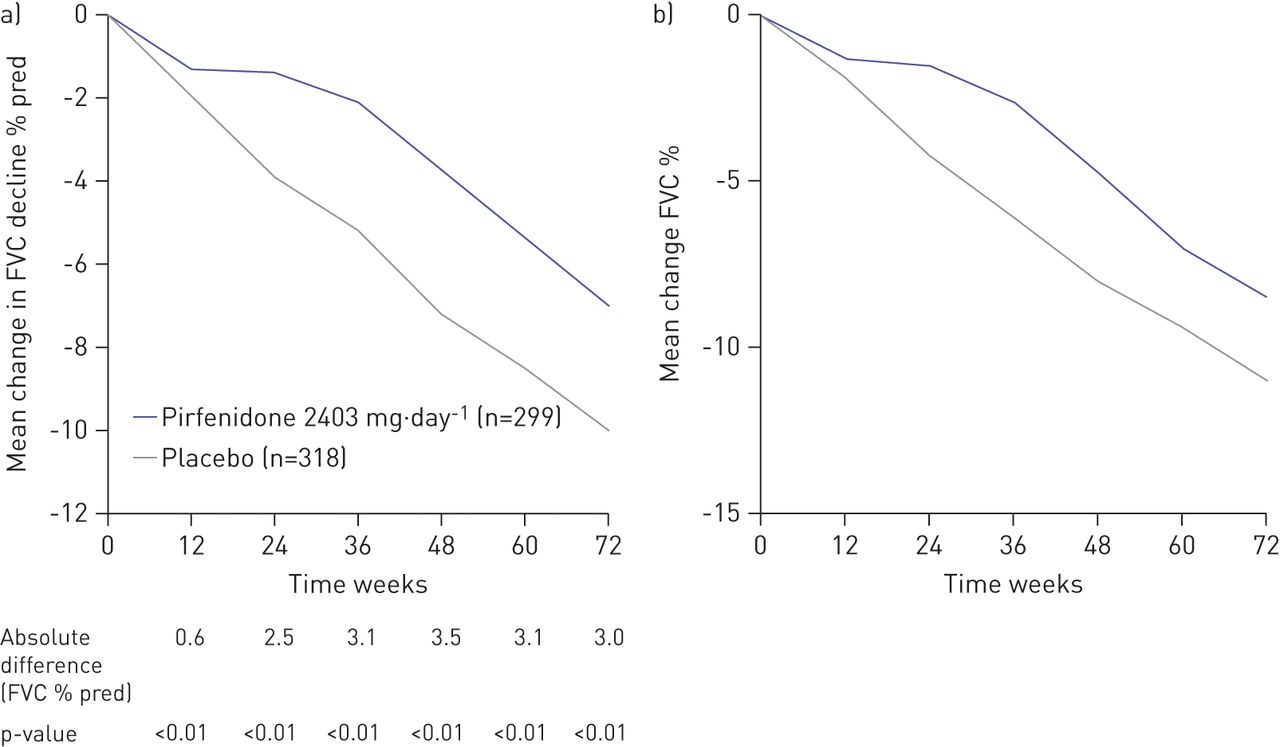
Mean change in forced vital capacity (FVC) decline from baseline to week 72 in a) the per-protocol (relative reduction 30%) and b) primary intention-to-treat (relative reduction 23%) analyses of the pooled population CAPACITY (Clinical Studies Assessing Pirfenidone in IPF: Research of Efficacy and Safety Outcomes) studies 004 and 006. b) Reproduced from [7] with permission from the publisher.
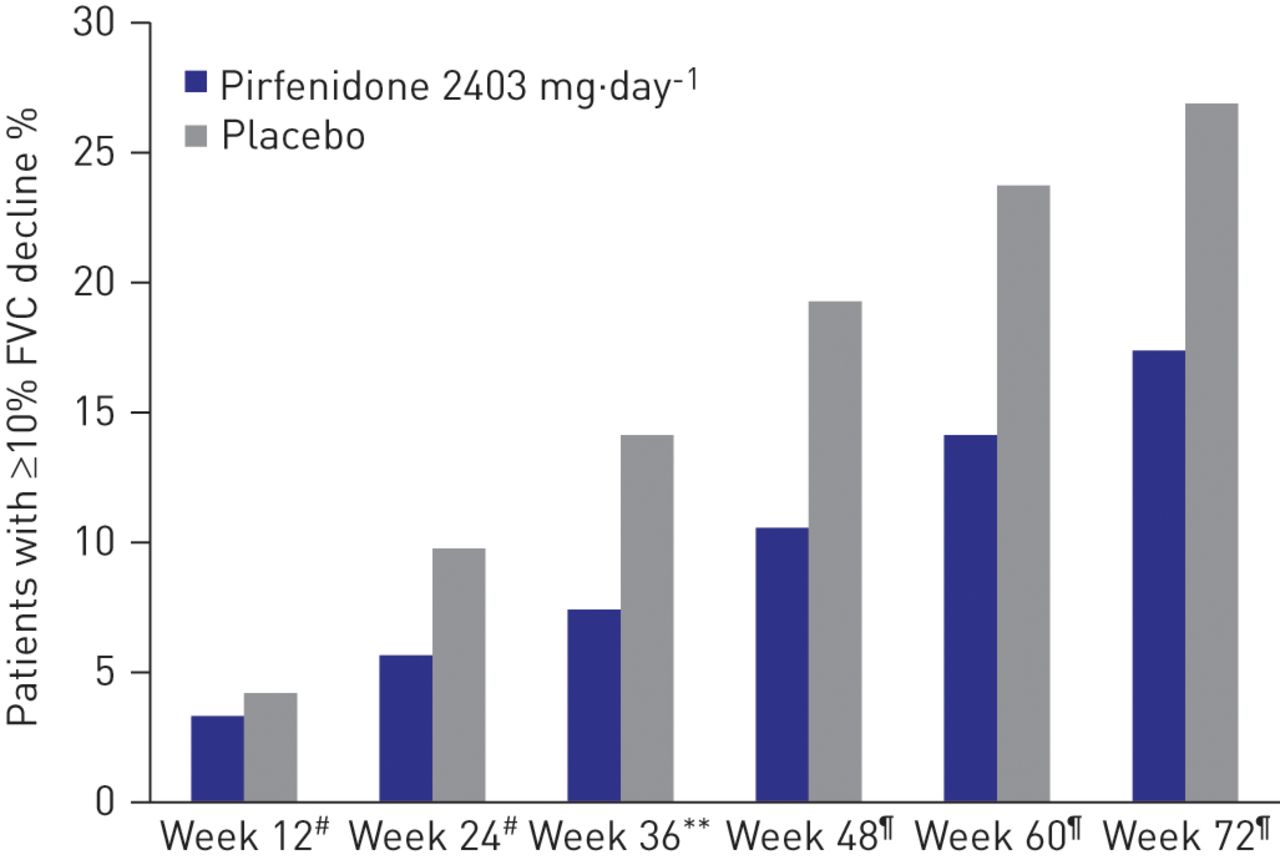
Patients with a ≥10% decline in forced vital capacity (FVC) or death in the per-protocol analysis of the pooled population CAPACITY (Clinical Studies Assessing Pirfenidone in IPF: Research of Efficacy and Safety Outcomes) studies 004 and 006. **: p<0.01. #: p = nonsignificant; ¶: p<0.005.
Although the CAPACITY study was not powered for mortality analysis, a pooled analysis of the exploratory end-point of mortality (ITT population) has been performed [7]. In the per-protocol analysis, all-cause mortality at week 72 was lower in the pirfenidone group (4.4%) compared to the placebo group (7.9%) (fig. 3). Similarly, in the ITT analysis, the hazard ratios for all-cause mortality (0.77, p = 0.315) and IPF-related mortality at any time during the study favoured pirfenidone over placebo (0.62, p = 0.117). However, other predictors of survival should also be considered.

All-cause mortality at week 72 in the per-protocol analysis of the pooled population CAPACITY (Clinical Studies Assessing Pirfenidone in IPF: Research of Efficacy and Safety Outcomes) studies 004 and 006. Relative reduction 44%. p = 0.3 (nonsignificant).
Efficacy of pirfenidone in CAPACITY patients with FVC decline at 6 months
Du Bois et al. [13] showed that an FVC decline of ≥10% over 24 weeks was a powerful predictor of mortality in the subsequent year for patients with IPF. In the overall CAPACITY population at week 24, 6.7% (n = 20) of patients treated with pirfenidone experienced a decline in FVC ≥10% compared with 11% (n = 28) of patients on placebo. An analysis was conducted in this subgroup to evaluate their subsequent clinical course and better understand clinical outcomes with pirfenidone after disease progression. In this subgroup of patients, 15% of patients who continued to receive pirfenidone subsequently experienced a further ≥10% decline in FVC at week 48 compared to 21.4% of patients receiving placebo. Additionally, a higher proportion of patients continuing treatment with pirfenidone (55%) experienced no decline in FVC at week 48 compared to 39.3% of patients receiving placebo (fig. 4). While the overall number of patients in this analysis was low and did not allow statistical analyses, a trend for more frequent disease stabilisation in patients treated with pirfenidone versus placebo was observed.

Outcomes at week 48 in patients from the CAPACITY (Clinical Studies Assessing Pirfenidone in IPF: Research of Efficacy and Safety Outcomes) pooled population with forced vital capacity (FVC) decline ≥10% at week 24 (occurring between week 24 and week 48). #: includes FVC decline ≥10%, death or lung transplantation.
Pirfenidone longer term experience
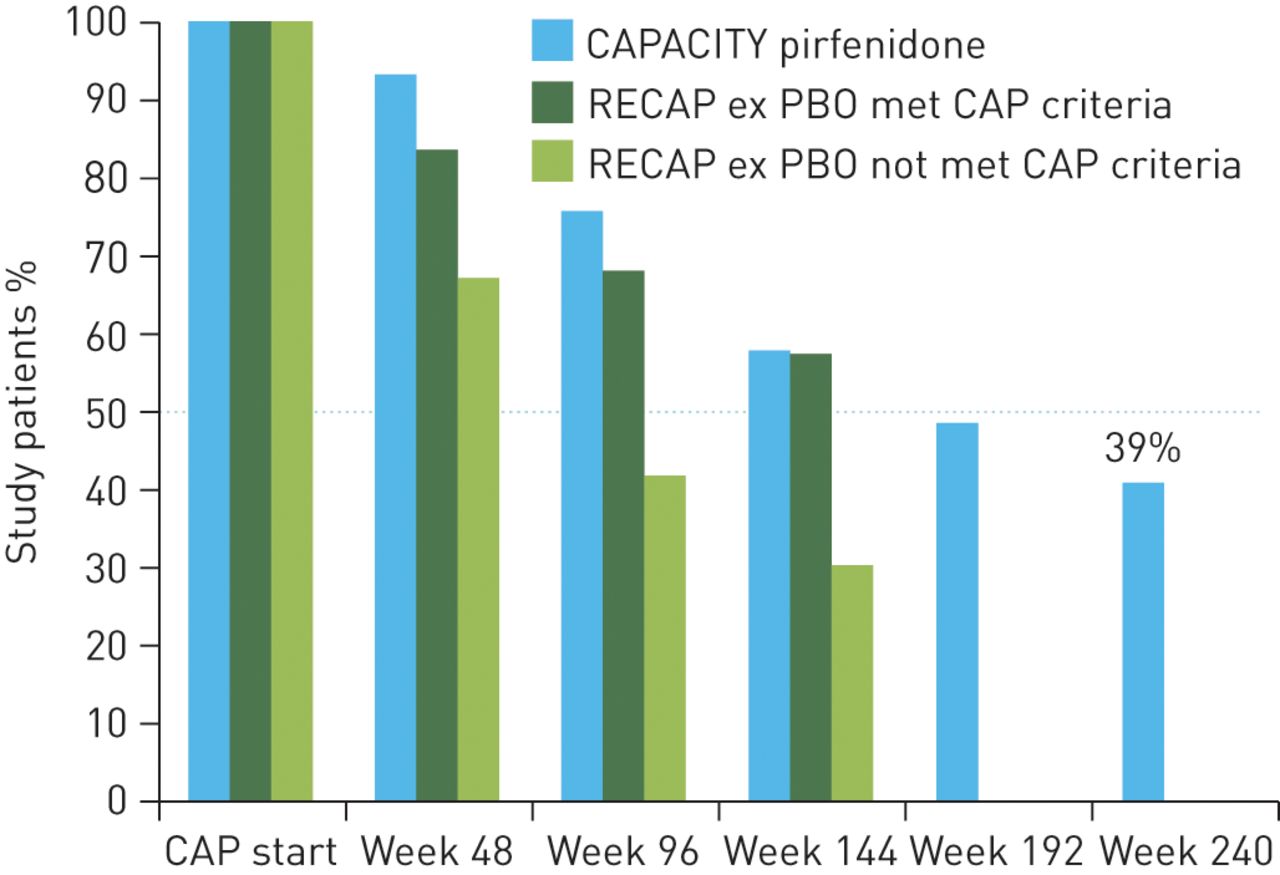
RECAP is an ongoing, open-label extension of the CAPACITY trial, which is collecting longer term data of pirfenidone in the treatment of IPF [14]. The data presented here are derived from an interim analysis with a data cut-off time-point of April 2012. Placebo-treated patients from the CAPACITY studies were switched to full-dose pirfenidone (2403 mg·day−1) while patients originally treated with pirfenidone continued with active treatment. A total of 603 patients (mean age 68.3 years, 72% male, mean 2.6 years since IPF diagnosis) were originally enrolled in this study [14]. Data from patients initially randomised to pirfenidone 2403 mg·day−1 in CAPACITY studies and subsequently included in RECAP had a follow-up time of almost 5 years (240 weeks) and demonstrated that ∼50% of patients who originally received pirfenidone in the CAPACITY studies were still alive and remained on treatment at almost 4 years (week 192) and ∼40% at week 240. Similar retention rates were also observed for patients who met the CAPACITY eligibility criteria when switching from placebo to pirfenidone in RECAP after 144 weeks of follow-up. However, discontinuation rates are higher for patients originally on placebo who did not meet the CAPACITY eligibility criteria when switching to active treatment (patients who may have had more severe impairment of lung function or longer duration of disease before enrolment) (fig. 5) [14]. The leading reasons for discontinuation of pirfenidone in CAPACITY and RECAP were adverse events (57.6% and 65.8% in CAPACITY and RECAP, respectively), patient decision (14.8% and 16.4%, respectively) and lung transplantation (10.3% and 4.9%, respectively). The analysis of RECAP suggests that pirfenidone is a suitable drug for long-term treatment of patients with mild-to-moderate IPF. However, the RECAP data must be interpreted with caution due to possible selection bias with regard to both pirfenidone (patients selected for tolerability and treatment response) and placebo (selection for mild progression because death or significant worsening led to informed drop out).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Study retention rates in the RECAP extension study. CAPACITY: Clinical Studies Assessing Pirfenidone in IPF: Research of Efficacy and Safety Outcomes; CAP: CAPACITY; ex PBO: placebo group in randomised placebo-controlled CAPACITY studies.
Pirfenidone single-centre experience
In addition to clinical trial analyses evaluating pirfenidone in patients reflective of the real-world setting, there is increasing data becoming available from everyday clinical practice.
Wijsenbeek et al. [15] retrospectively reported efficacy and tolerability with pirfenidone in a cohort of 55 IPF patients encountered in clinical practice. Follow-up information was available in eight patients at 12 months, 15 patients at 9 months and 27 patients at 6 months, with stabilisation of FVC % pred observed in the majority. In a subgroup of patients (n = 19), a clinical cough score (1 = no cough; 10 = worst cough) was recorded over 6 months. In 11 patients the cough score decreased after 1 month of pirfenidone treatment (mean reduction -2.2 points), remained unchanged in seven patients and increased in one patient. This data supports the reported efficacy and tolerability profile in a real-world setting but also provides information on the potential effect of pirfenidone on improving cough symptoms, which is interesting since severe coughing is known to have a significant impact on patient quality of life [15]. An observational study of the effect of pirfenidone on cough and quality of life in patients with IPF in daily practice is currently ongoing [16].
In another single centre cohort from a German tertiary referral centre for interstitial lung diseases, Oltmanns et al. [17] evaluated pirfenidone in everyday clinical practice in 63 patients. While patient characteristics were reflective of a “typical” IPF clinical cohort, in the majority of patients treatment with pirfenidone was initiated later in the disease than in the published RCTs. In addition, these patients had a high percentage of relevant comorbidities (e.g. 44% coronary artery disease and 17% emphysema), which are often excluded from clinical trials. Analysis of patients who had pulmonary function data available 3–12 months before and ≥6 months after starting pirfenidone therapy (n = 17), showed a reduced FVC decline and disease stabilisation in the majority of these patients (72%). As expected, pirfenidone-related side-effects were common but mostly manageable with supportive care [17].
Conclusion
Evolving data from clinical trials and experience in everyday practice is helping to further support the clinical profile of pirfenidone in adult patients with mild-to-moderate IPF. Sub-analyses of the CAPACITY studies and data from the RECAP extension study might suggest that the benefits of pirfenidone can be maximised, particularly in patients at greater risk of FVC decline. A per-protocol population is, notwithstanding potential bias, perhaps more reflective of the treatment benefit for patients who continue with pirfenidone treatment, and the CAPACITY per-protocol population analysis compared favourably with the CAPACITY ITT population across primary and secondary end-points, as well as mortality which was pre-specified as an exploratory end-point, providing further confidence in the robustness of the original registration studies. Data from patients completing the CAPACITY studies who continued on treatment in RECAP also highlights the positive longer term continuation rates on treatment.
Increasingly, new data is emerging on the use of pirfenidone in everyday clinical practice and supports the benefits of pirfenidone in the real-world setting. This clinical experience should also help clinicians to identify potential adverse events with pirfenidone. To proactively help clinicians managing pirfenidone-related adverse events, an expert panel discussion on recommendations based on existing guidelines, research evidence and consensus opinions will be published in the future, focusing mainly on preventing and/or mitigating gastrointestinal and skin-related adverse events. These include taking pirfenidone during (or after) a meal, avoiding sun exposure, wearing protective clothing, and applying a broad-spectrum sunscreen with high UVA and UVB protection.
Acknowledgments
This article is based on the proceedings of a satellite symposium held at the 2013 European Respiratory Society Annual Congress (Barcelona, Spain), which was sponsored by InterMune. I was assisted in the preparation of the article by IntraMed International (Milan, Italy), funded by InterMune.
Footnotes
Provenance: Publication of this peer-reviewed article was supported by InterMune International AG, Muttenz, Switzerland (article sponsor, European Respiratory Review issue 131).
Statement of Interest: Disclosures can be found alongside the online version of this article at err.ersjournals.com
- Received December 6, 2013.
- Accepted January 8, 2014.
- ©ERS 2014
ERR articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 3.0.
References









