





Abstract
Fibrocytes are bone marrow-derived, circulating mesenchymal progenitor cells that play a role in several fibrotic disorders, including lung fibrosis. They are attracted to injured tissue by various chemokines. It is likely that fibrocytes play a detrimental role in tissue homeostasis and promote fibrosis, although this paradigm needs further confirmation. This would make fibrocytes a possible novel treatment target for fibrotic disorders. Fibrocytes also have some potential as a biomarker for idiopathic pulmonary fibrosis (IPF) and other diseases, but the promising preliminary data from single centre studies still require independent validation. Despite several, as yet, unresolved issues, it has become clear that fibrocytes are more than an incidental finding in lung injury and repair, and may hold great promise for the future of IPF management.
Introduction
Fibrosis is a common biological phenomenon in health and disease. It can affect all organ systems of the body. Fibrosis is defined by the overgrowth, hardening and/or scarring of tissues and is attributed to excess deposition of extracellular matrix (ECM), including collagens, proteoglycans and glycoproteins [1]. Controlled synthesis, deposition and degradation of ECM is a physiological process in tissue repair. In contrast, abnormal ECM accumulation is a critical part of the pathogenesis of several common conditions in the lung, including chronic infectious airway, vascular and parenchymal diseases [2]. There are many factors that can contribute to the switch from normal, regulated ECM production and degradation to excessive or aberrant deposition in fibrosis. Many of these factors are a major area of study in fibrosis, in order to better understand the process and develop novel strategies to manage patients with fibrotic disorders. Fibrocytes are circulating bone-marrow derived mesenchymal cell progenitors that can differentiate into fibroblasts and myofibroblasts once they enter the tissue. They are a putative source for fibroblastic foci, which are a characteristic feature of the histopathological pattern “usual interstitial pneumonia” found in idiopathic pulmonary fibrosis (IPF). The exact biological role of fibrocytes in tissue repair and fibrosis is not fully understood. They are believed to be involved in the disease pathogenesis of several fibrotic disorders affecting the lungs, liver, kidneys and other organs, and may have a role as a potential biomarker of disease activity as they can be detected and quantified from easily available peripheral blood samples.
Pulmonary fibrosis
Pulmonary fibrosis can be the result of many known diseases including systemic disease (e.g. scleroderma or mixed connective tissue disease), occupational diseases (such as asbestosis and silicosis) and drug-related lung disorders among others. The most progressive and unpredictable type of lung fibrosis is IPF [3]. This disease is a major clinical problem, particularly in the elderly population. Numerous epidemiological studies have suggested a role for certain contributing factors but, to date, the initiating cause of IPF is still unknown [4]. Patients with IPF suffer from worsening dyspnoea and cough and can experience various patterns of progression during the disease course, eventually resulting in irreversible lung damage and decline of lung function [5]. This process often leads to respiratory failure with a median survival of 3 years after diagnosis [6, 7].
There are many challenges remaining in the management of IPF patients despite major advances in the past decade and the approval of the first IPF-specific drug, pirfenidone, in most countries (except the USA) [8]. The detailed understanding of the pathogenesis will help to delineate disease mechanisms that can be targeted by novel therapeutic compounds [9]. Further study of disease mechanisms can also help to detect new biomarkers, which are required to measure disease activity and treatment responses, and estimate prognosis in individual IPF patients. One of the key questions is to identify the factors that turn tissue repair away from normal healing and towards limited, non-progressive fibrosis (e.g. as is often seen in asbestosis or idiopathic nonspecific interstitial pneumonia) or even progressive fibrosis, as seen in IPF. It is possible that the fundamental biological mechanisms may not be substantially different between these scenarios. Alteration of the ECM composition and the microenvironment in which structural cells live in the organ tissue is an increasingly recognised problem and a potential driver of fibrogenesis in IPF [10].
There is a need for a closer study of both local (within the lung) and systemic responses (from the circulation and bone marrow) in fibrotic lung disease. Bone marrow-derived progenitor cells and systemic responses to signals that originate in damaged tissue are an important factor not only in host defence, but also in tissue repair. Better understanding of their contributions to injury might also allow us to identify unifying principles that could be present in different fibrotic disorders.
Progenitor cells and fibrocytes in tissue repair and fibrosis
There is increasing evidence suggesting that tissue-resident and bone marrow-derived progenitor cells contribute to injury repair in the lungs, including epithelial, mesenchymal and endothelial cell progenitors [2, 11, 12]. Small niches of progenitor cells resident in the lungs are involved in repair and re-epithelialisation after injury [13]. It is believed that in diseases of exaggerated repair, such as IPF, the depletion of “beneficial” progenitor cells may lead to increased recruitment of alternate sources of progenitors, leading to a switch from sucessful repair to fibrotic remodelling. Ageing is an important factor in this process as well, which is consistent with the fact that IPF typically affects individuals >60–65 years of age. Animal models have shown that old mice primarily mobilise fibrocytes from the bone marrow in response to bleomycin-induced lung injury. In contrast, mesenchymal stem cells are the predominant cell type found in the circulation of young mice following administration of bleomycin. Consequently, young mice develop less severe fibrosis in the lungs compared to older mice [14]. Interestingly, mesenchymal stem cells are more pluripotent than fibrocytes, providing a possible explanation for the difference in the wound healing process between age groups [15].
Fibrocytes are spindle-shaped mesenchymal progenitor cells, and were first characterised by Bucala et al. [16]. Since then many disease models have shown that these cells respond to tissue-derived signals and migrate to sites of injury where they can differentiate into fibroblast-like cells [12, 17]. Fibrocytes circulate in the peripheral blood and are capable of producing collagen, other ECM components, crosslinking enzymes, cytokines and growth factors. Fibrocytes express a variety of mesenchymal markers including collagen 1, as well as the leukocyte marker CD45 and the haematopoietic stem cell marker CD34 [18]. Different groups have reported many different marker sets as being useful in identifying fibrocytes. These have been extensively reviewed by Pilling et al. [19]. However, there is sufficient evidence that these cells can be reliably identified in the circulation by markers for CD45 and Col-1 [20–23].
Fibrocytes respond to the profibrotic cytokine transforming growth factor (TGF)-β by expressing α-smooth muscle actin, which supports a potential role of these cells in myofibroblast formation, a prototypical feature of lung fibrosis [24]. In pulmonary fibrosis, the abnormal tissue remodelling is characterised by excessive accumulation of ECM, distortion of lung architecture including the vascular system [25], and formation of fibroblastic foci. These foci represent the sites of active fibrogenesis, and fibrocytes are a supposed source of cells that create these fibroblast clusters. Besides fibrocytes, the proliferation of resident fibroblasts, epithelial to mesenchymal transition [26] and pericytes [27] are potential contributors to fibroblast foci (fig. 1). All of these cells, when exposed to TGF-β and other local factors, may undergo transformation into myofibroblasts [28], which then drives progressive fibrogenesis [6, 29]. Interestingly, there is increasing evidence that not only can cytokines switch progenitor cells into active myofibroblasts, but they also cause abnormal matrix composition and rigidity [10]. It has been shown that fibrocytes demonstrate phenotype plasticity and are affected by their niche environment [30, 31]. Careful dissection and creation of accurate models of the fibrotic matrix are required to truly understand the functions of fibrocytes in the microenvironments of different organs.
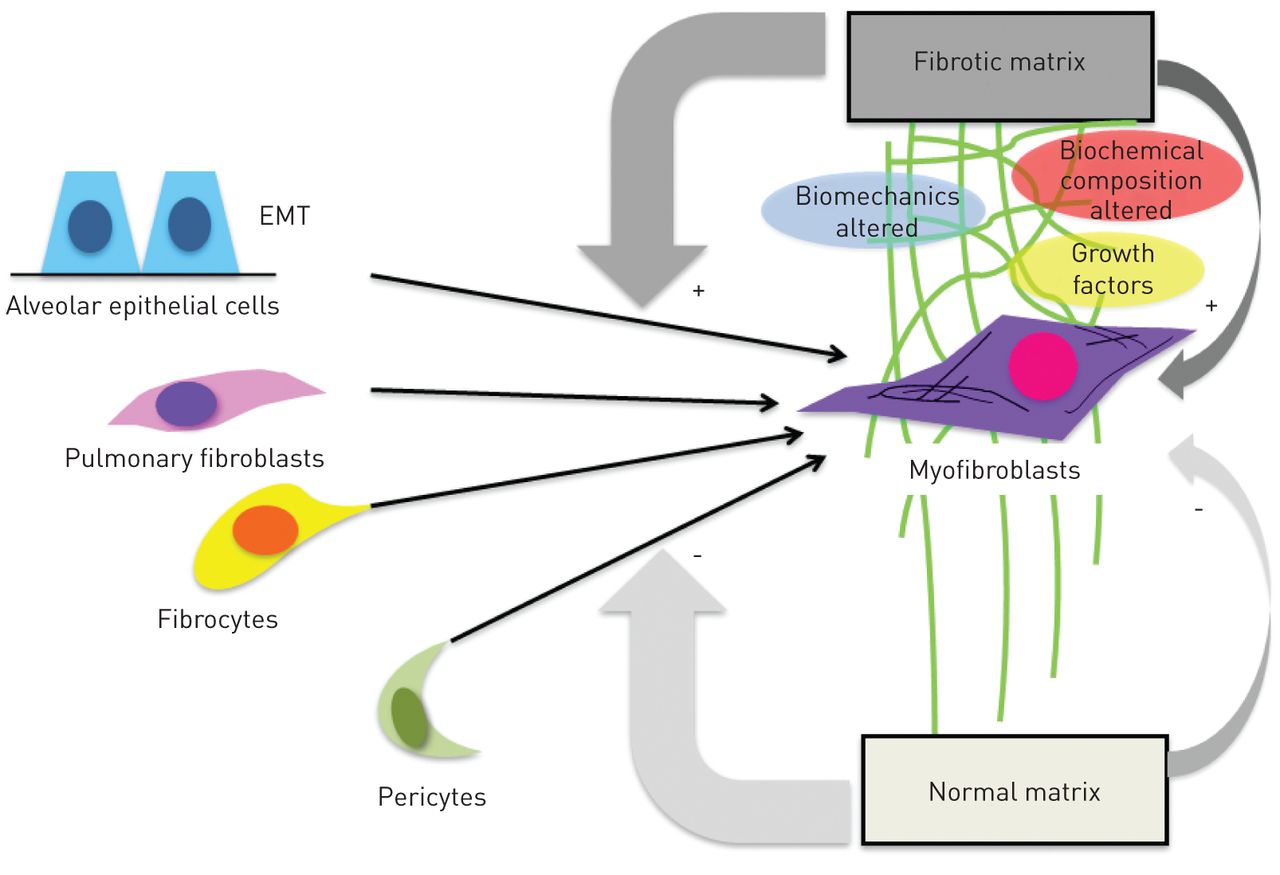
{kind=link}
Proposed cellular sources for fibroblast foci and the fibrotic matrix. Several fibroblast progenitor cells, particularly fibrocytes, alveolar epithelial cells (via epithelial to mesenchymal transition (EMT)), resident fibroblasts and pericytes, can differentiate into myofibroblasts and thereby contribute to fibroblast focus formation. It is possible that the altered biochemical composition of fibrotic matrix has a profound impact on this differentiation process and facilitates the accumulation and persistence of fibrogenic cell phenotypes.
Fibrocytes have been found in patients with various fibrotic disorders and in animal models of injury [12, 32, 33], suggesting a relationship between mesenchymal cell progenitors and chronic injury and repair. There is evidence that these cells are involved in the pathogenesis of lung fibrosis [34], liver fibrosis [35], scleroderma [36, 37] and asthma [38, 39]. Fibrocytes have been identified in areas of injury adjacent to newly formed scar tissue, which has raised the question of whether or not these cells are good targets to improve repair and prevent fibrotic remodelling. It is also still unclear which chemokines mobilise fibrocytes from the bone marrow and recruit them to the sites of tissue injury. Amongst them are human CCL2 [12], CXCL12 [17] and secondary lymphoid chemokine [24, 40] (table 1). There is increasing support for the hypothesis that fibrocytes differentiate into myofibroblasts, as has been demonstrated in vivo [33, 38, 41] and in vitro [24]. Serum amyloid P appears to be an inhibitor of fibrocyte differentiation and is currently being developed for IPF treatment [42]. More knowledge about fibrocyte biology in vivo is still needed before they can be confirmed as a major treatment target. However, the concept is supported by studies which have clearly shown that increased numbers of fibrocytes are worse for lung injury [21, 32].
Fibrocytes as a prospective biomarker
The clinical management of IPF remains a major challenge not only due to limited drugs being available to treat the disease, but also because of the lack of good indicators for disease progression. The identification of suitable biomarkers is an important task; these biomarkers would help act as predictors of prognosis and possibly treatment response [43]. The National Institutes of Health defines a biomarker as “any characteristic that can be objectively measured and act as an indicator for the different processes that occur in the human body” [44]. An ideal biomarker must be objectively measured, reproducible, easily detected and inexpensive. High sensitivity and specificity combined with the ability to predict clinical changes in disease are key properties that candidate biomarkers should fulfil [45]. Exciting developments in molecular biology have opened up new approaches for biomarker discovery, but circulating blood cells remain an attractive target. For fibrotic disorders, these include circulating fibrocytes.
There have been several important studies investigating the potential of fibrocytes as a biomarker of disease activity in fibrotic lung disease (table 2) [20–23, 43, 46, 47]. Work by our group [21], Strieter et al. [29], LaPar et al. [23], and Fujiwara et al. [22] has shown that circulating fibrocytes may have the ability to predict the progression of lung fibrosis. In our study, the survival of IPF patients with circulating fibrocytes >5% of total blood leukocytes was 7.5 months compared with 27 months for patients with <5% fibrocytes [21]. Importantly, the predictive value of fibrocytes was independent of other outcomes, such as forced vital capacity or diffusing capacity of the lungs for carbon monoxide. This observation was recently confirmed in a smaller, single centre study in Japan [22]. Another study showed a correlation between the numbers of circulating fibrocytes and the development of bronchiolitis obliterans syndrome after lung transplantation [23]. Before these promising, yet preliminary, studies can assist in the management of IPF patients or find a use as outcomes for clinical trials, they have to be confirmed and validated in larger multicentre trials. Some of these important studies are currently recruiting and will hopefully provide some answers to these open questions in the near future [43].
Conclusion
Fibrocytes are bone marrow-derived, circulating mesenchymal progenitor cells that play a role in several fibrotic disorders, including IPF. They are attracted by tissue-derived chemokines, which are yet to be confirmed. It is likely that they play a detrimental role in tissue homeostasis and promote fibrosis, although even this paradigm needs further confirmation. Whether or not fibrocytes are attractive treatment targets in IPF remains to be proven. Fibrocytes may have some predictive value as biomarkers for IPF and other diseases, but the promising preliminary data still requires independent and large scale validation. Several major challenges remain, including a poor understanding of fibrocyte variability in health and disease and the fact that many researchers use different panels of markers to identify fibrocytes. Some consistency would be useful to put the vast amount of information in the fibrocyte literature into a common context. Despite these issues, it has become clear that mesenchymal cells and their progenitors are more than incidental findings in lung injury and repair and hold promise for the future.
Footnotes
Conflict of interest: Disclosures can be found alongside the online version of this article at err.ersjournals.com
Provenance: Submitted article, peer reviewed.
- Received October 10, 2013.
- Accepted October 21, 2013.
- ©ERS 2013
ERR articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 3.0.
References









