





Abstract
Idiopathic pulmonary fibrosis (IPF), the most prevalent of the idiopathic interstitial pneumonias, is associated with a poor prognosis. An accurate diagnosis of IPF is essential for its optimal management. The 2011 American Thoracic Society (ATS)/European Respiratory Society (ERS)/Japanese Respiratory Society (JRS)/Latin American Thoracic Association (ALAT) recommendations on the diagnosis and management of IPF were developed from a systematic review of the published literature. High-resolution computed tomography (HRCT) scanning has a central role in the IPF diagnostic pathway, with formal designation of criteria for an HRCT pattern of usual interstitial pneumonia. In the correct clinical context, a usual interstitial pneumonia pattern on HRCT is indicative of a definite diagnosis of IPF and negates the need for a surgical lung biopsy. However, although the 2011 ATS/ERS/JRS/ALAT statement is a major advance, the application of the guideline recommendations by clinicians has identified limitations that should be addressed in future statements. Key problems include: 1) HRCT misdiagnosis, particularly by less experienced radiologists; 2) lack of management recommendations for the highly prevalent clinical scenarios of “probable” or “possible” IPF; 3) ongoing confusion concerning the diagnostic role of bronchoalveolar lavage; and 4) the lack of integration of clinical data in the designation of the diagnostic likelihood of IPF, including the treated course of disease. These issues become evident as the recommendations are applied and highlight the need for continued guideline adjustments.
- Bronchoalveolar lavage
- diagnosis
- guidelines
- high-resolution computed tomography
- idiopathic pulmonary fibrosis
- surgical lung biopsy
Idiopathic interstitial pneumonia (IIP) makes up a heterogeneous group of diseases incorporated collectively under the umbrella term of interstitial lung diseases (ILDs) [1]. In 2002, a multidisciplinary panel from the American Thoracic Society (ATS) and the European Respiratory Society (ERS) proposed a classification of IIPs that comprises pathological entities including idiopathic pulmonary fibrosis (IPF), nonspecific interstitial pneumonia (NSIP), respiratory bronchiolitis-associated ILD, cryptogenic organising pneumonia, acute interstitial pneumonia, desquamative interstitial pneumonia and lymphoid interstitial pneumonia (fig. 1) [1].
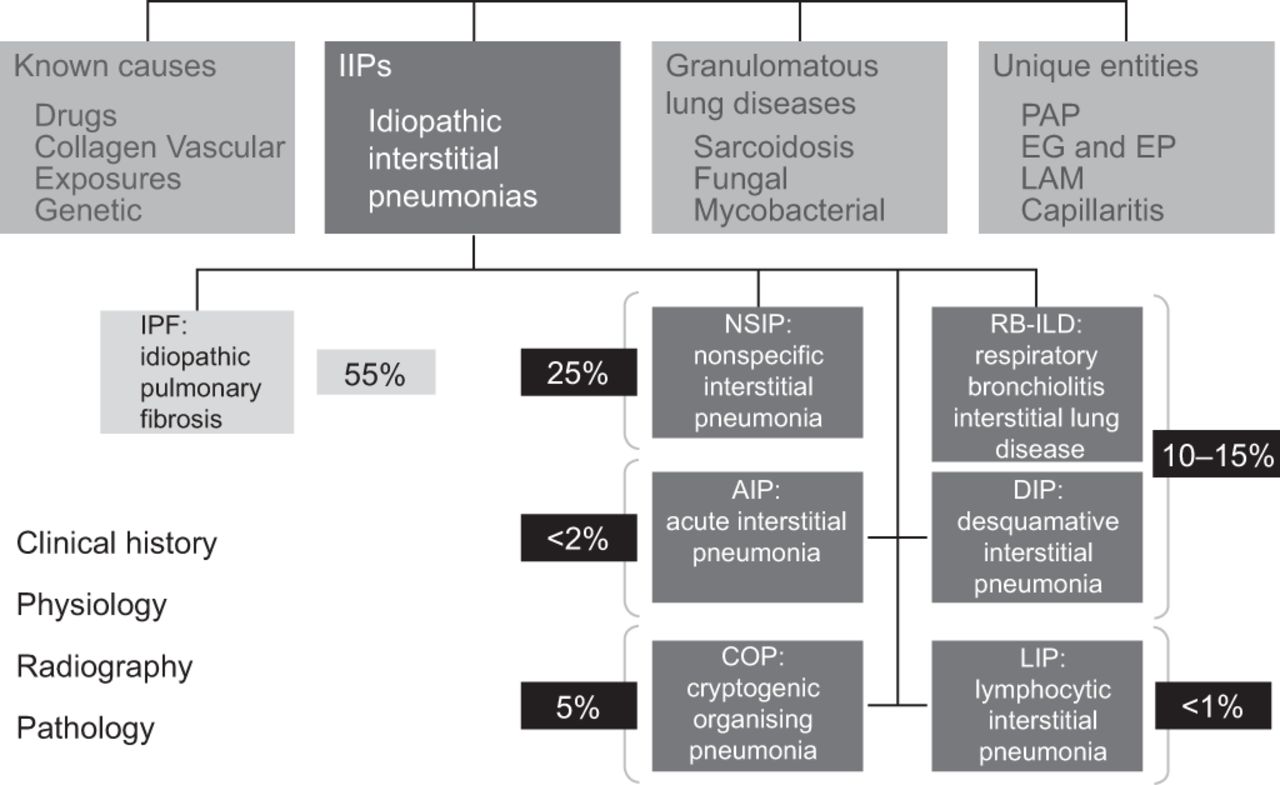
Classification of interstitial lung disease. EG: eosinophilic granulomatosis; EP: eosinophilic pneumonia; LAM: lymphangioleiomyomatosis. Reproduced and modified from [1] with permission from the publisher.
IPF is defined as a specific form of chronic, progressively fibrosing IIP that occurs primarily in relatively elderly adults. It is the most pernicious and frequent of the ILDs, accounting for 55% of IIP [1–3]. The prevalence of IPF in the European Union is ∼120 000 [4, 5], and an estimated 40 000 new cases are diagnosed each year [6]. IPF is associated with a characteristic radiological and/or histopathological pattern of usual interstitial pneumonia (UIP) [1]. A histologically confirmed UIP pattern of IPF is associated with a significantly worse prognosis than that of other histological patterns of chronic interstitial pneumonia [6, 7] and carries an estimated 5-year survival rate that is worse than several types of cancer [6, 8]. table 1 shows the clinical conditions associated with UIP pattern [1].
An early and accurate diagnosis of IPF is therefore critical, particularly with the advent of novel specific treatments that may have the potential to reduce disease progression [9]. However, establishing an accurate diagnosis of IPF is challenging and requires an integrated multidisciplinary approach involving pulmonologists, radiologists and pathologists [6]. This article considers the development, merits and potential limitations of the current diagnostic recommendations for IPF. In particular, the practicalities and interpretation of recent advances in high-resolution computerised tomography (HRCT), and the appropriateness of surgical lung biopsy (SLB) and/or bronchoalveolar lavage (BAL) in making a differential diagnosis of IPF in routine clinical practice are considered.
EVOLUTION OF DIAGNOSTIC CRITERIA
In 2000, the ATS and ERS published the first consensus statement outlining guidelines for the diagnosis and treatment of IPF [10]. This statement identified four major and four minor diagnostic criteria for IPF that together identified a pattern of UIP (table 2) [10]. For example, the absence of known causes of lung fibrosis, including collagen vascular disease, drug toxicity, and various environmental exposures, represents a key factor in the diagnostic process.
However, there are significant limitations concerning the four minor criteria of these diagnostic guidelines. For example, the specification of patient age of ≥50 years excludes a small but key patient group in which an early diagnosis is particularly desirable. Further, a slow onset of disease does not acknowledge the fact that some IPF patients initially present with acute symptomatic exacerbations. Similarly, it is also difficult to define and accurately distinguish IPF from the symptomatic course of patients with a disease duration of ≥3 months with co-existing pulmonary fibrosis and/or pre-existing smoking-related lung damage. Moreover, the presence of crackles on auscultation is a highly nonspecific diagnostic sign of IPF.
Revised guidelines
The 2000 diagnostic and management guidelines for IPF were updated in 2011 by an international collaboration of the ATS and ERS, as well as the Japanese Respiratory Society (JRS) and the Latin American Thoracic Association (ALAT) [11]. The main objective was to provide simplified and evidence-based criteria that facilitate a more confident clinical diagnosis of IPF. The recommendations were developed from a thorough review of existing published evidence, with the use of expert opinion only when the evidence base was inadequate.
The original 2000 diagnostic criteria proposed that, in an appropriate clinical context, an UIP pattern according to HRCT criteria is sufficient to diagnose IPF without performing a SLB [1, 10]. This proposition was ratified by the revised ATS/ERS/JRS/ALAT 2011 guidelines that established HRCT as having a primary role in the IPF diagnostic pathway. The revised guidelines specifically identify the HRCT features of IPF “UIP”, “possible UIP” and “inconsistent with UIP” patterns (table 3) [11]. The summary of the revised diagnostic criteria is as follows: 1) exclusion of other known causes of ILD (e.g. domestic and occupational environmental exposures, connective tissue disease and drug toxicity); 2) presence of a UIP pattern on HRCT scans in individuals not subjected to SLB; and 3) specific combinations of HRCT and SLB pattern in individuals subjected to SLB.
The HRCT imaging features of UIP are characterised by reticular opacities, often associated with traction bronchiectasis, honeycombing manifested as mainly subpleural cluster cystic airspaces, typically of comparable diameters (3–10 mm, but occasionally larger), with well-defined thickened walls, and the absence of additional radiological features considered incompatible with a diagnosis of IPF [9, 11]. Ground-glass opacities are common, but less extensive than the reticulation, with a characteristically basal and peripheral distribution, though often patchy [2, 3].
IMPLICATIONS FOR CLINICAL PRACTICE
The ATS/ERS/JRS/ALAT 2011 management guidelines for IPF represent an impressive review of the literature and a disciplined approach to a previously undisciplined field. The designation of “definite”, “probable” or “possible” IPF is a major advance over the previous guidelines. However, the revised 2011 recommendations remain focused on the management of patients with a “definite” diagnosis of IPF and are not easily applied when the diagnosis of IPF is less certain. Patients with probable or possible IPF are frequently encountered in clinical practice and often have a differential diagnosis of fibrotic (NSIP) or chronic hypersensitivity pneumonitis [11].
The presence or absence of honeycombing
The principle of integrating HRCT with histopathological data in assigning a diagnostic likelihood of IPF is a significant improvement of the 2011 recommendations. Indeed, the strengths and potential drawbacks of the revised IPF guidelines centre largely upon the application and analysis of HRCT. However, one of the most prevalent problems in clinical practice is the classification of patients with a predominantly basal and subpleural distribution of reticular abnormalities typical of IPF, but without honeycombing on HRCT. Atypical HRCT findings include ground-glass opacity, pleural effusion and thickening with nodules and/or scattered cysts. Unfortunately, there is no category in the 2011 guidelines for this constellation of features that are not uncommonly seen in patients with possible IPF in clinical practice, including those with traction bronchiectasis, co-existing emphysema and fibrosis [12], and/or those of ≥60 years of age [9].
The accuracy of a trained observer in discriminating between ILDs and the identification of IPF is approximately 80–90% [13–15]. However, interobserver variation in the distinction between typical and atypical HRCT appearances of IPF is substantial among less experienced observers [16]. Expert interpretation of HRCT data and the identification of honeycombing are therefore central to the diagnosis of IPF [17]. Unfortunately, however, the current HRCT diagnostic recommendations are not implemented uniformly by the majority of radiologists. Thus, additional diagnostic evaluation is often required in patients with possible IPF that have evidence of peripheral basal reticular change in the absence of honeycombing with other atypical HRCT findings.
Surgical lung biopsy
As suggested above, a diagnosis of definite IPF is not usually possible from HRCT appearances alone in at least one-third of cases [3, 18]. As suggested in the 2000 guidelines, the revised 2011 guidelines appropriately recommend that a SLB should be performed when patients present with nondiagnostic scans or if the diagnosis of definite IPF is uncertain [2, 11]. However, a large proportion of patients with suspected IPF cannot undergo a SLB procedure, either because of contraindications (e.g. comorbid conditions, disease severity, advanced age, etc.) or because the patient declines the procedure [19]. Such patients are therefore disenfranchised by the diagnostic criteria and consequently ineligible from receiving novel therapies licensed for use in definite IPF. A clearer separation between patients who can undergo a SLB with an acceptably low risk and those in whom SLB should be avoided would be helpful to clinicians.
Bronchoalveolar lavage
The value of BAL in the diagnosis of IPF is also contentious. The most important application of BAL is to increase the index of suspicion for alternative disorders in patients with suspected IPF, including hypersensitivity pneumonitis and NSIP, by the demonstration of a lymphocytosis (>30%) [20]. The ATS/ERS/JRS/ALAT 2011 guidelines make a weak negative recommendation that BAL cellular analysis should not be performed in the diagnostic evaluation of IPF in the majority of patients, but may be appropriate in a minority of patients [11].
This recommendation is confusing as it may be applied to either of two very dissimilar clinical scenarios. First, if HRCT features are classical for IPF and the clinical context is correct, the patient has definite IPF. Secondly, if the HRCT features are not classical for IPF but IPF is suspected, the diagnostic overlap includes IPF/NSIP, IPF/hypersensitivity penumonitis, IPF/sarcoidosis and IPF/unclassifiable. The combination of such scenarios in a single recommendation is clinically impractical and two independent statements would be preferable. In the British Thoracic Society guidelines on the diagnosis and management of interstitial lung disease, for example, the diagnostic use of BAL is reserved for patients in whom the diagnosis of IPF is uncertain after clinical assessment and HRCT scanning, with the case-by-case exclusion of patients with severe disease, or because a biopsy will ultimately be performed [21].
Rational multidisciplinary clinical approach
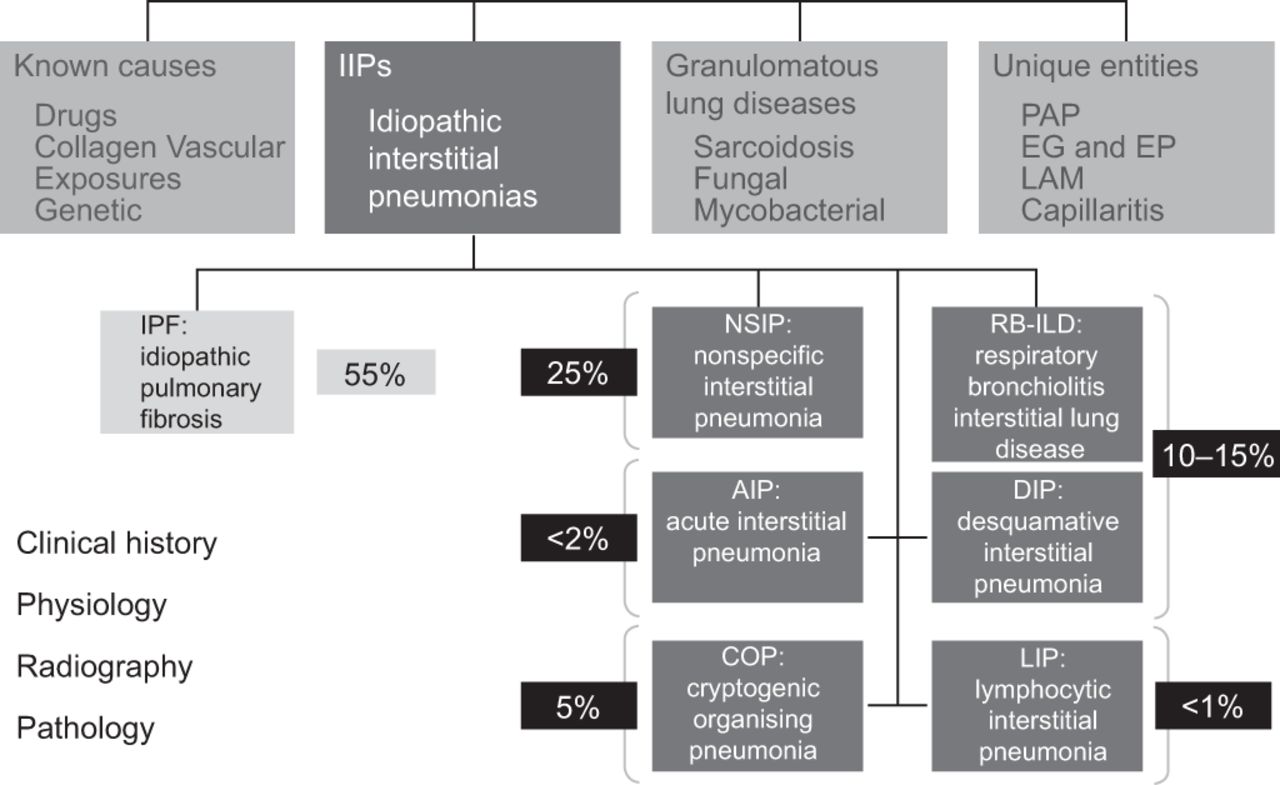
The ATS/ERS/JRS/ALAT 2011 guideline recommendations have assisted clinicians in making an evidence-based diagnosis of IPF (fig. 2) [11]. However, no attempt is made to define minimum standards required for the process of a multidisciplinary evaluation of IPF, particularly with respect to HRCT misdiagnosis. A multidisciplinary, dynamic approach within an expert centre and regular collaborative input of clinicians, radiologists, and histopathologists has been shown to improve diagnostic accuracy [22–24]. For example, and where relevant, consultation with the surgeon, pathologist and radiologist in interpreting BAL results, and to determine the exact location(s) of surgical biopsy based on HRCT appearances is advantageous [11]. A nurse specialist should also attend this meeting to oversee and integrate the process.

{kind=link}
{kind=link}
2011 American Thoracic Society/European Respiratory Society/Japanese Respiratory Society/Latin American Thoracic Association diagnostic algorithm for idiopathic pulmonary fibrosis (IPF). ILD: interstitial lung disease; HRCT: high-resolution computed tomography; UIP: usual interstitial pneumonia; MDD: multidisciplinary discussion. Reproduced and modified from [11] with permission from the publisher.
DISCUSSION
While commendable and a considerable improvement upon the original 2000 recommendations, some aspects of the most recent 2011 ATS/ERS/JRS/ALAT diagnostic guidelines clearly merit review. For example, the revised guidance focuses mainly on definite IPF, most often diagnosed from typical HRCT appearances. Diagnostic uncertainties, partly due to the multiple different ways in which physicians approach IPF (e.g. the availability of appropriate lung biopsy specimens and accurate medical histories), along with the variability in the natural history of disease and HRCT appearances, all contribute to the inherent confusion surrounding this topic. In addition, the BAL recommendation has caused confusion due to the use of a single recommendation to cover two radically different clinical scenarios.
Clearly, training in applying guideline recommendations in the diagnosis of IPF should be directed not only to radiologists but also to pathologists and clinicians. As with any other disease, these issues highlight the continued need for updating the guidelines, including diagnostic and therapeutic aspects, based upon the limitations that become evident as recommendations are applied. An ERS consensus statement is now under way to address these and other problems.
Footnotes
Provenance
Publication of this peer-reviewed article was supported by InterMune Inc, USA. (principal sponsor, European Respiratory Review issue 128).
Statement of Interest
Conflict of interest information can be found alongside the online version of this article at err.ersjournals.com
- Received March 6, 2013.
- Accepted March 26, 2013.
- ©ERS 2013
REFERENCES









