





Idiopathic pulmonary fibrosis (IPF) is the most prevalent of the idiopathic interstitial pneumonias and its incidence seems to be increasing [1–4]. IPF is generally considered to be unresponsive to “standard” therapies and carries a poor prognosis with most patients dying within at least 5 years following diagnosis [5–7]. An accurate diagnosis of IPF is essential for the optimal management of patients with IPF. The original IPF management guidelines, published in 2000 [8], have been superseded recently by the evidence-based international recommendations jointly sponsored by the European Respiratory Society (ERS), the American Thoracic Association (ATS), the Japanese Respiratory Society (JRS) and the Latin American Thoracic Association (ALAT) [9]. Nonetheless, despite this and other major accomplishments over the last decade, the diagnosis and the management of IPF remain a major medical challenge. Diagnostic uncertainties remain due, at least in part, to variability in the natural history of the disease and co-existing complicating comorbidities, interpretation and widespread application of the guideline recommendations, and analysis of histological and radiographic assays, as well as a paucity of accurate indicators of disease progression and response to treatment.
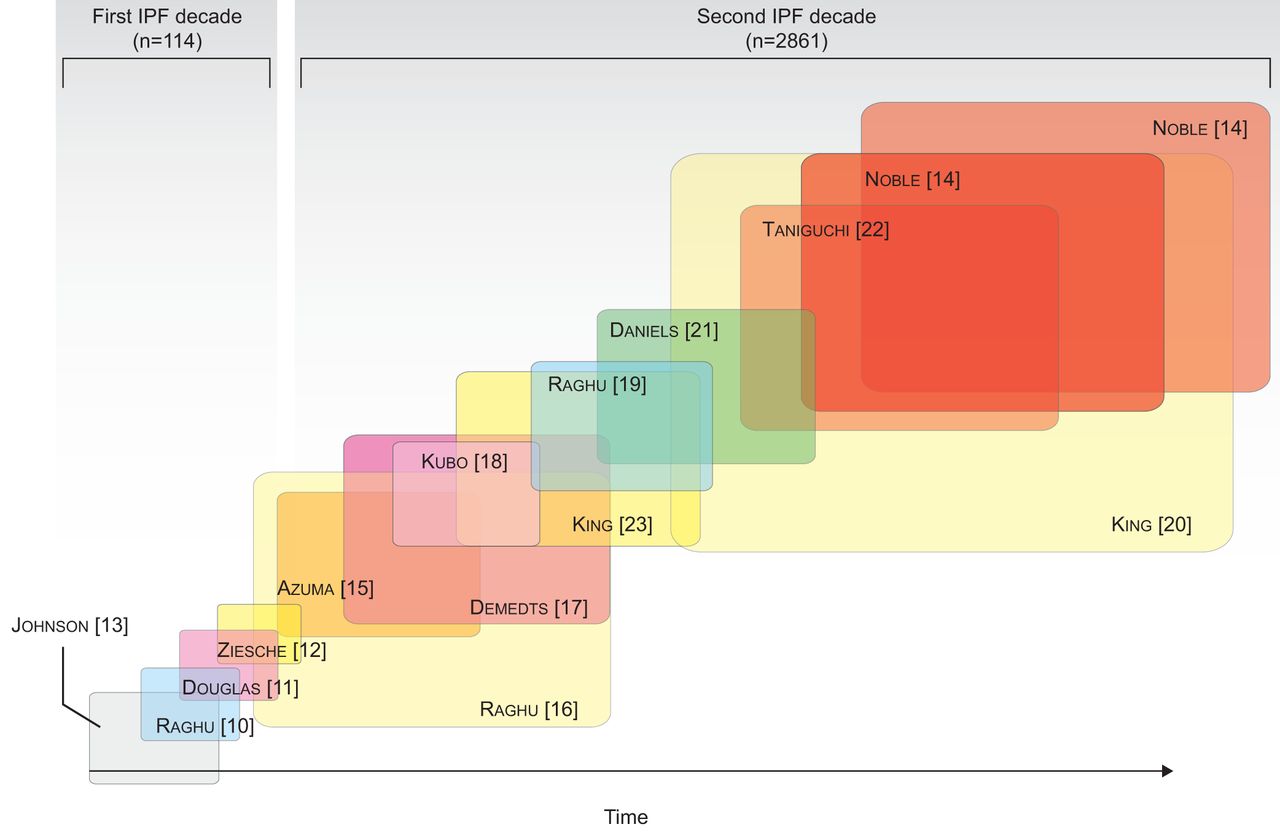
Since a cure for IPF is not currently available, the treatment goal is to stabilise or reduce the rate of disease progression. There have been significant and encouraging increases in the number, size and quality of randomised clinical trials in IPF over the past decade (fig. 1) [10–23], and novel anti-fibrotic agents with the potential to realise this treatment goal are now starting to emerge. For example, although the 2011 guidelines committee did not recommend any single pharmacological agent, pirfenidone has been approved for clinical use in Europe.
{kind=link}
The identification of more accurate and objective predictors of clinical status, prognosis and survival estimates in IPF is also critical for clinical management and individual treatment decision making [24]. There is also an ongoing debate on the choice of primary end-point for use in clinical trials of treatments for IPF. Improvements in molecular techniques have expanded our understanding of IPF and have led to the identification of new molecular pathways, thus shaping the potential for a more targeted therapeutic approach [25, 26]. While genetic studies have provided some insights into the pathogenesis of IPF, more functional studies that confirm their significance and studies investigating other mutations, associations and gene–environment relationships are required [27–30].
This issue of the European Respiratory Review includes a series of state-of-the-art articles based on presentations by prominent IPF experts at the recent 2012 ERS Annual Congress in Vienna, Austria, and the 17th International Colloquium on Lung and Airways Fibrosis (ICLAF) held in Modena, Italy. The articles review the substantial progress that has been made in the pathogenesis, diagnosis, management and treatment of IPF, but also anticipate the future advances in the understanding and clinical management of IPF. In the first article, Wells [31] provides a critical review and the latest insights into managing diagnostic procedures in IPF and identifies limitations that should be addressed in future consensus statements. Behr [32] then provides an updated evaluation of clinical study and evidence-based data of novel treatment strategies in IPF, including the latest results from the pirfenidone CAPACITY (Clinical Studies Assessing Pirfenidone in Idiopathic Pulmonary Fibrosis: Research of Efficacy and Safety Outcomes) clinical trials and the subsequent open-label extension study, RECAP (Open-label roll-over study from CAPACITY). Maher [33] goes on to identify the most recent potential biomarkers and disease fingerprint of IPF and describes the ongoing PROFILEing (Prospective Observation of Fibrosis in the Lung Clinical Endpoints) Study, an innovative UK-based multicentre prospective cohort study that aims to characterise composite biological and clinical trial end-points. Finally, Cottin [34] describes the impact of a new syndrome, in which pulmonary fibrosis co-exists with pulmonary emphysema. Combined pulmonary fibrosis and emphysema (CPFE) is a strong determinant of secondary pulmonary hypertension and further studies are needed to ascertain the aetiology, morbidity, mortality and management of CPFE, with or without pulmonary hypertension, and to evaluate novel therapeutic options.
The expert contributions in this series of articles indicate that there is room for cautious optimism in this challenging era of respiratory medicine, and that we may be entering an era in which prevention of IPF progression may become a reasonable therapeutic goal. Recently updated guidelines have provided important recommendations for the diagnosis and pharmacological and non-pharmacological management of patients with IPF. The ability of physicians to achieve a confident diagnosis has greatly improved, and the proliferation of clinical trials has started to build an evidence base upon which rational treatment decisions can be built.
Acknowledgments
I received medical writing support from IntraMed International (Milan, Italy), which was funded by InterMune Inc. (Brisbane, CA, USA).
Footnotes
Provenance
Publication of this peer-reviewed article was supported by InterMune Inc., USA (principal sponsor, European Respiratory Review issue 128).
Statement of Interest
Conflict of interest information can be found alongside the online version of this article at err.ersjournals.com
- ©ERS 2013
REFERENCES









