





Abstract
Thymic epithelial tumours represent a wide range of anatomical, clinical, histological and molecular malignant entities that may be aggressive and difficult to treat. The histopathological classification distinguishes thymomas from thymic carcinomas. Thymomas may be associated with autoimmune disorders.
The management of thymic epithelial tumours is a paradigm of co-operation between clinicians, surgeons and pathologists, from establishing the diagnosis to organising the multimodal therapeutic strategy. Surgery is the mainstay of the curative-intent treatment, as complete resection represents the most significantly favourable prognostic factor on overall survival. In case of invasion of intra-thoracic structures and/or dissemination to the pleura and the pericardium, precluding complete resection to be achieved, primary chemotherapy has been used to reduce the tumour burden, possibly allowing subsequent surgery and/or radiotherapy.
Novel strategies are needed, especially for refractory, recurrent tumours and thymic carcinomas, which carry a poor prognosis. Personalised approaches are currently being developed, as potentially “druggable” molecular targets are emerging from recent integrated genomic analyses. Along with the large variety of questions relative to the treatment strategy, thymic epithelial tumours represent a model of therapeutic implementation and achievement in orphan thoracic oncology, showing how the advent of new results induces new questions, as well as diversifies further clinical research directions and international collaborative initiatives.
Thymic epithelial tumours represent a wide range of anatomical, clinical, histological and molecular entities that may be aggressive and difficult to treat [1–4]. The World Health Organization (WHO) histopathological classification distinguishes thymomas from thymic carcinomas. Thymomas are further subdivided into different types (A, AB, B1, B2 and B3) based on the morphology of epithelial tumour cells (with an increasing degree of atypia from type A to B3), the relative proportion of the non-tumoural lymphocytic component (decreasing from type B1 to B3) and resemblance to normal thymic architecture (table 1) [3]. Thymic carcinomas are similar to their extra-thymic counterpart, with the most frequent subtype being squamous cell carcinoma. Neuroendocrine tumours will not be discussed in this review. Tumour invasiveness, as evaluated by the Masaoka staging system, is a major predictor of outcome [5, 6]. The WHO classification is correlated with stage at diagnosis, as 80–90% of type A to B1 thymomas are stage I–II tumours, whereas 50–60% of type B2 and 60–80% of type B3 thymomas and carcinomas present with stage III–IV extent, a unique feature which may explain its reported prognostic value [6–9]. Given a higher propensity toward nodal and systemic invasiveness, tumour, node, metastases (TNM)-based staging may be more appropriate for thymic carcinomas [3, 4, 10].
The physiological role of the thymus gland is to induce central tolerance to self-antigens, through the control of the differentiation and the subsequent positive and negative selection of immature T-lymphocytes [11]. In particular, the medullary thymic epithelial cells have the unique capability to express tissue-related antigens in the context of the major histocompatibility complex; self-reactive thymocytes are signalled to die via programmed cell death, and thereby deleted from the immune repertoire. The ectopic expression of self-antigens is regulated by the transcription factor AIRE (autoimmune regulator) [12]. AIRE is under-expressed in thymomas, leading to a lack of negative selection towards key peripheral proteins and, consequently, to the development of autoimmune disorders, the most frequent being myasthenia gravis, hypogammaglobulinaemia and pure red cell aplasia [13]. Myasthenia gravis is caused by circulating antibodies that block acetylcholine receptors at the post-synaptic neuromuscular junction, and is found in 30% of patients with thymoma, when 15% of patients presenting with myasthenia do have thymoma [14]. Systematic immunological check-up is recommended when a diagnosis of thymoma is suspected, including complete blood count including reticulocytes and serum protein electrophoresis, as well as anti-acetylcholine receptor and antinuclear antibodies tests. Autoimmune so called “para-thymic” disorders have been reported to significantly affect the overall survival of patients, especially in early-stage tumours [15]. It is estimated that about 25% of patients with thymic epithelial tumour actually die from autoimmune disease-related complications; of note, another 25% die from thymoma-unrelated causes. Overall survival may then be a debatable end-point for prognostic studies and time-to-progression may be preferred [15]. Treatment of parathymic syndromes has to be conducted in parallel with the oncologic management; in particular, control of myasthenia gravis-related symptoms must be obtained before surgery. Contrary to cytokine-related paraneoplastic syndromes, the treatment of the thymic tumour does not systematically improve autoimmune manifestation; the appearance or recurrence of autoimmune disease does not predict recurrence of the thymic tumour [14].
The management of thymic epithelial tumours is a paradigm of co-operation between clinicians, surgeons and pathologists, from establishing the diagnosis to organising the therapeutic strategy and evaluating the prognosis. Surgery is the mainstay of the curative-intent treatment of thymic tumours [16–20], as complete resection represents the most significant favourable prognostic factor, both on disease-free and overall survival [6, 7, 21]. However, nearly 30% of patients present with locally advanced tumour at the time of diagnosis, with invasion of intra-thoracic neighbouring structures, and/or dissemination to the pleura and the pericardium, precluding upfront surgery to be performed in a carcinologic setting. In such cases, chemotherapy has been used both to reduce the tumour burden, possibly allowing subsequent surgery and/or radiotherapy, and to achieve prolonged disease control [17–20]. For invasive tumours (Masaoka stage II and beyond), the need for post-operative radiotherapy is highly debated. Chemotherapy is also a palliative-intent treatment of unresectable, metastatic and recurrent tumours, which are rare in thymoma but more frequent in thymic carcinomas [22, 23].
As a consequence of their rarity, our knowledge about the management of patients with thymic epithelial tumours has mainly been based upon retrospective series, uncontrolled non-randomised trials or expert opinion. Recently, the International Thymic Malignancy Interest Group (ITMIG) published consensual guidelines and definitions, so that consistent approaches may be adopted worldwide, especially in the setting of clinical research, to clarify areas of uncertainty [24]. Refinement of the Masaoka staging system represents a major achievement in this setting (table 2) [24]. The definition of major and minor criteria for the WHO-based histopathological diagnosis of thymic epithelial tumours represent another major challenge, currently being addressed by ITMIG, as inter-observer reproducibility for the classification of thymomas has been reported to be as low as 40–60%, given the frequent heterogenity of large tumours and the occurrence of combined tumours in approximately 15–20% of cases [25].
Novel strategies are needed, especially for refractory, recurrent tumours and thymic carcinomas, which carry a poor prognosis despite multimodal treatment. Over recent years, significant efforts have been conducted to dissect the molecular pathways involved in thymic epithelial cells carcinogenesis [26]. Potentially druggable targets are emerging, laying the foundations to implement personalised medicine for patients.
OVERVIEW OF TREATMENT STRATEGY
The treatment strategy for thymic epithelial tumour is primarily based on whether the tumour may be resected upfront or not, as complete resection represents the most significant and consistent prognostic factor on disease-free and overall survival [6, 9, 16, 21]. As no clinical staging system has been developed so far, the evaluation of resectability is mainly based on the surgeon’s expertise. Pre-operative computed tomography (CT) findings associated with tumour invasiveness were identified in a series of 99 patients from the MD Anderson Cancer Center (Houston, TX, USA) [27]; the most significant variables associated with stage III/IV disease, less likely to be resectable upfront at diagnosis, were a tumour size >7 cm (OR 3.18, 95% CI 1.16–8.67; p=0.02), lobulated contour (OR 8.20, 95% CI 1.63–41.35; p=0.010) and infiltration of surrounding fat (OR 3.76, 95% CI 1.45–9.78; p=0.007). In an ongoing trial evaluating several modalities for pre-operative treatment, inclusion criteria predicting unresectability include a tumour size >8 cm, or a size ranging from 5 to 8 cm with one or more of the following criteria: multifocal calcification, heterogeneous appearance, irregular or scalloped borders, great vessel invasion or encirclement. For tumours <5 cm in greatest diameter, obvious great vessel invasion/encirclement has to be present on the CT scan (Clinicaltrial.gov NCT00387868). The value of a positron emission tomography (PET) scan to predict histology, hyperplasia versus tumour, thymoma versus thymic carcinoma or thymoma subtype, or invasiveness has been debated [28, 29]. PET may be of interest for subsequent follow-up of patients, to detect recurrences after first-line treatment [29]. After surgery, post-operative radiotherapy may be delivered.
If complete resection is deemed to be achievable upfront, surgery represents the first step of the treatment; if not, primary chemotherapy is administered as part of the curative-intent sequential strategy integrating subsequent surgery or radiotherapy [2, 17, 23]. In this later situation, histopathological diagnosis has to be obtained through small biopsy. In general, tumours located in the anterior mediastinum are approached using percutaneous core needle biopsy, mediastinotomy or mini-thoracotomy [30]. Fine-needle aspiration may not be recommended, as cytological specimens of thymic tumours may be hard to interpret. Pleural spaces should not be punctured to avoid tumour cell seeding. Most frequent differential diagnoses include thymic hyperplasia, as well as lung cancer and other primary mediastinal malignancies, such as lymphoma or germ-cell tumours. An extensive review of these entities is out of the scope of this review.
Chemotherapy may also be delivered as palliative-intent treatment in advanced, non-resectable, non-irradiable or metastatic thymic tumour to improve tumour-related symptoms.
SURGERY PRINCIPLES
As a standard, the surgical management of thymic epithelial tumours requires the wide opening of the mediastinum and both pleural cavities, which is classically achieved by median sternotomy [2, 31, 32]. Generally, complete thymectomy, including the tumour, the residual thymus and perithymic fat, is preferred because local recurrences have been observed after partial thymectomy. The first step of the operation consists of a careful examination of the mediastinum and pleural cavities, followed by evaluation of macroscopic capsular invasion, infiltration of peri-thymic and mediastinal fat, peritumoral and pleural adherences, and involvement of surrounding tissues. These findings, together with the subsequent pathological examination of the surgical specimen, constitute the basis of staging (table 2) [5, 24]. If the tumour is invasive, en bloc removal of all affected structures, including lung parenchyma (usually through limited resection), great vessels, phrenic nerves and pleural implants, should be performed. Thoracotomy may be mandatory. Areas of uncertain margins are marked with clips to allow precise delivery of post-operative radiotherapy. Phrenic preservation may be balanced with complete resection in patients with severe myasthenia gravis. Frozen sections to assess tumour involvement of resection margins are not recommended, given the high risk of false negative results [31].
Over the past decade, minimally invasive surgery of the thymus has been developed, including a variety of access incisions, methods of exposure, visualisation and equipment [33, 34]. This includes transcervical, extended transcervical, video-assisted thoracoscopy and robotic approaches (right or left, right and left, right and cervical, left and cervical, subxiphoid and right and left, cervical and subxiphoid). In a retrospective comparative analysis, operative time was higher with the robotic approach, but hospital stay was shorter and neurological outcome seemed to be better [35]. The choice for minimally invasive resection should not degrade or change the principles that are deemed appropriate for an open approach, including: resection of the tumour, the thymus and the mediastinal fat; dissection and visualisation of the innominate vein and both phrenic nerves; sufficiently large access incision to prevent specimen disruption; use of a retrival bag; and exploration of the pleura. Conversion to open surgery is mandatory if required to achieve complete resection, and is not considered a complication of the minimal approach.
The incidence of lymph node metastases is largely undetermined in thymic epithelial tumours, as it is unclear in the reported literature how often nodes are biopsied or examined [31]. In the landmark Japanese series of 1,320 resected thymic tumours, lymph node invasion was mostly located in the anterior mediastinum, and was found in 2% of thymomas, including <1% of stage I cases, and 6% of stage III cases [6]. Nodal invasion is higher in thymic carcinomas, as it occurs in the anterior mediastinum in 70% of cases, in other intrathoracic locations in 35% of cases, and in extrathoracic sites in 30% of cases [4, 6]. The unfavourable prognostic value of nodal tumour invasion is only established for carcinomas [6]. Any suspicious nodes (e.g. enlarged, firm or hypermetabolic at PET-scan) should actually be removed and separately labelled and submitted. Routine removal of anterior mediastinal nodes is recommended for stage III–IV thymomas; in this situation, a systematic sampling of intrathoracic sites is encouraged (i.e. paratracheal, aortopulmonary window and subcarinal areas depending on tumour location) [31]. An even more extensive nodal dissection, including anterior mediastinal, intrathoracic, supraclavicular and lower cervical areas, may be discussed in thymic carcinoma.
Hyperthermic intrapleural chemotherapy, as well as extrapleural pneumonectomy, may be discussed in case of stage IVA tumour [36, 37]. Decision-making criteria are still unclear.
SURGICAL PATHOLOGY RECOMMENDATIONS
Recent recommendations to facilitate the surgical pathology diagnosis of thymic epithelial tumours were recently made by ITMIG [31]. Communication between surgeons and pathologists is crucial at this step. The proper orientation of the specimen and the designation of involved structures, organs or areas of concern are the primary responsibility of the operating surgeon, and may be prepared using a mediastinal board, consisting of a line diagram of the mediastinum placed on a simple cork or wax board. The operative note should mention the following elements: whether gross tumour was left behind and, if so, location of residual tumour; extent of resection performed; presence and location of any adhesions that were simply divided (not suspicious for involvement); any additional structures or organs removed; any sites of intra-operative concern, including how these were marked on the specimen and in the patient; which nodal areas were explored and the extent of assessment; and the presence or absence of pleural and pericardial lesion.
The final pathological examination of each specimen lead to a final histological diagnosis and staging of the tumour based on the WHO classification and the refined Masaoka–Koga system, respectively (tables 1 and 2) [3, 24]. Given the potential heterogeneity of thymic epithelial tumours, at least five representative sections should be examined regardless of the tumour diameter; one additional block should be taken per additional centimetre. Capsular integrity or invasion should be explicitly mentioned. Completeness of resection has to be assessed making the distinction between tissues that have been cut or dissected, and a surface bounded by a space (such the mediastinal pleura, pericardium or endothelium of the innominate veins) that should not be designated as a positive margin. A tumour extending up to inked margin in an area of tissue disruption that was identified as not grossly concerning intra-operatively, may not correspond to invaded margin. In all situations in which the margin is <3 mm, the distance separating the tumour from the closest inked margin of resection should be documented; if the margin is <1 mm at least three additional levels through this area should be obtained [31].
RADIOTHERAPY PRINCIPLES
Radiotherapy has been used in two distinct settings for the treatment of thymic epithelial tumours: 1) it may be administered after surgical resection, aiming at decreasing the risk of mediastinal recurrence; and 2) it may also be used as definite treatment in locally advanced tumours that are deemed to remain unresectable after primary chemotherapy [38, 39]. Current modalities of radiotherapy for thymoma include the following. 1) The use of multi-field arrangement conformal radiotherapy and three-dimensional treatment planning. 2) A clinical target volume including the whole thymic space, the tumour and its extensions, and the anterior, superior and middle mediastinum (with reduction of fields after a total dose of 50–55 Gy). Prophylactic supraclavicular nodes irradiation is no longer recommended, as isolated recurrences in this area are exceptional. 3) A total dose ranging from 40 to 60 Gy, including a boost on the tumour bed in incompletely or non-resected lesions, as mentioned previously, surgical clips may be then useful to plan the gross tumour volume, with a standard-fractionation scheme consisting of daily doses ranging from 1.8 to 2 Gy over a 4–6-week period. Standard dose constraints for thoracic radiotherapy should be used. Although thymomas have been recognised as highly radiosensitive for years, the benefit of dose escalation on local control has not clearly been established. Arriagada et al. [40] reported similar local control rates with total doses inferior to 48 Gy or superior to 60 Gy, while in more recent series, no improvement regarding local control was observed with doses of 50 Gy or >60 Gy [41]. Ultimately, higher doses are usually delivered, if possible, in the setting of definite treatment, as compared to that of post-operative treatment, with total doses of 40–50 Gy and 50–60 Gy with a boost up to 60–66 Gy, respectively. Patients with thymic tumour may have a prolonged survival and, if so, late-onset radiation-induced toxicities have to be considered. In-field malignancies may occur. Moreover, radiotherapy may be useful for the treatment of subsequent tumour recurrences [42].
Post-operative radiotherapy
Post-operative radiation is typically administered within 2 months after surgery. When the tumour has been completely resected, minimum fields for post-operative radiation should encompass the pre-operative extent of disease, as indicated by pre-operative imaging and regions of risk identified intra-operatively (“involved field” treatment). The field may then encompass involved nodes and the site of a resected pleural implant. The role of post-operative radiotherapy remains highly debated. The low number and the delay of recurrences make statistical analyses underpowered in the majority of studies. More than tumour stage, completeness of surgical resection is such a significant factor of overall survival that the intrinsic weight of radiotherapy is hard to evaluate [21].
Masaoka stage I thymomas carry a recurrence rate after complete resection that is as low as 1–2% with a 5-yr survival of 100% after exclusive surgery [5, 21]. Post-operative treatment is then highly unlikely to demonstrate any significant improvement. A Chinese trial including 29 patients compared adjuvant radiotherapy versus exclusive surgery in patients with completely resected stage I thymomas, and, although underpowered, failed to show any differences in patient outcome [43]. Furthermore, a recent report from the Survival, Epidemiology, and End Results (SEER) database including 275 stage I thymic tumours suggested a possible adverse impact of post-operative radiotherapy on 5-yr cause-specific survival (91% versus 98% without radiotherapy; p=0.03) [44]. Adjuvant radiotherapy is then rarely administered in stage I thymomas after complete surgical resection (fig. 1) [45].

Computed tomography scan of a 35-yr-old male who presented with chest pain, showing a mass in the anterior mediastium (arrow). Surgical resection was performed immediately. The tumour was a type AB stage I thymoma. No post-operative treatment was administered.
For stage II tumours, complete resection is achieved in 75–90% of cases, with 5-yr local control achieved in >85% of cases [6, 46–48]. In the landmark Japanese cohort including 257 stage II thymomas, no significant difference was observed regarding recurrence rates in patients who received surgery alone or surgery followed by adjuvant radiotherapy [6]. However, more recent series, involving the use of modern radiation delivery techniques, suggested post-operative radiotherapy might improve disease-free and overall survival even more in these patients; by up to 100% and 95%, respectively [41, 48–51]. Differences in outcome may exist depending on the extent of capsular invasion, i.e. whether tumour stage is IIa or IIb. In a SEER database study including 1,334 patients, post-operative radiotherapy resulted in a marginally significant survival benefit (p=0.09) [52]. In a recent study from the University of Pennsylvania (Philadelphia, PA, USA), this benefit was estimated to be 8%, despite being based on a small number of patients [53].
In completely resected stage III tumours, recurrence rates have been reported for a long time as being lower after post-operative radiotherapy [46, 54–56], ranging from 0% to 20% versus 40% to 55% after adjuvant radiotherapy or exclusive surgery, respectively [57–61]. Current practice is actually highly variable, but the global trend over recent years is clearly moving towards a less frequent use of post-operative radiotherapy, reserving it for high-risk cases. Moreover, it has become clear that recurrences occur outside the mediastinum in >60% of cases, especially in stage II–III tumours. A SEER database analysis included 626 invasive thymic tumours, and showed that 5-year survival was significantly better after post-operative radiotherapy (76% versus 66%; p=0.01), but cause-specific survival was similar (91% versus 86%; p=0.12) [44]. This finding may be potentially related to the high late toxicity rates of radiotherapy before the conformal era. Another SEER analysis on 1,464 patients confirmed these results [62]. However, in those analyses, no benefit from radiotherapy was observed after “extirpative”, i.e. R0 or R1, resection. A pooled analysis of retrospective studies comparing exclusive R0 surgery versus R0 surgery followed by radiotherapy for stage II–III thymomas and published from 1981 to 2008, was conducted by Korst et al. [63]. Overall, 592 patients from 13 studies were included: 250 who received adjuvant radiotherapy and 342 who did not. No significant difference was observed in the two groups (OR 1.05, 95% CI 0.63–1.75; p=0.63).
Integration of histology in the decision-making process for post-operative radiotherapy is complex. In a series of 79 patients with completely resected stage III-type B thymoma, only five presented with tumour recurrence [64]. In a Japanese series of 324 patients with resected thymic tumour, 119 of whom had received post-operative radiotherapy, specific survival was lower after radiotherapy in case of type A-AB-B1 tumour, while no survival difference was observed in case of type B2-B3 tumours, independently from tumour stage [65]. In another series of 107 patients with resected stage II tumours, type B3 histology was an independent unfavourable prognostic factor, suggesting the potential interest for radiotherapy in this subset of patients [66]. Data for thymic carcinoma are hard to interpret given the rarity and inconsistency in the diagnostic criteria. In the Japanese series, 186 patients with thymic carcinoma were analysed with no survival difference based on post-operative treatment, regardless of the tumour stage [6]. In a more recent series of 26 patients with thymic carcinoma treated with surgery and radiotherapy, local control rate was 82% higher than that observed in a historical series [67].
Based on these data, a proposal of recommendations for post-operative radiotherapy following complete resection of a thymic tumour is presented in table 3. Post-operative radiotherapy may be administered in the case of microscopically incomplete (R1) resection, with a boost to doses of at least 60 Gy on areas of concern. Radiotherapy consistently decreased recurrence rates from 60–80% to 21–45% in this setting [6, 50, 54, 55, 60, 61].
Hemithoracic radiotherapy, recently made feasible with the availability of intensity-modulated and tomotherapy techniques, has been hypothesised to be of interest in controlling the high risk of recurrence that occurs in the pleura [68]. Several reports indicate the feasibility and efficacy of delivering a 10–15 Gy prophylactic hemithoracic irradiation after complete resection and adjuvant radiotherapy, with a reduction of recurrence rates from 90–100% to 60–75%, without a clear effect on overall survival [68–70].
Definite radiotherapy
Definite radiotherapy is defined as radiotherapy delivered as the sole local modality for control of disease, although it is often combined with primary chemotherapy for systemic treatment [38]. Radiotherapy is used in case of a thymic tumour which remains unresectable or inoperable after induction treatment. Definite radiotherapy also refers to radiotherapy delivered when a macroscopically incomplete (R2) resection was previously performed. Thus, radiotherapy is used as a primary local modality for curative intent, with or without chemotherapy. This is because the role of debulking both on local control and recurrence-free survival remains uncertain, although it may facilitate further curative-intent radiotherapy through the reduction of the gross tumour volume [58, 71]. Treatment field guidelines should be delineated and reported as described previously. Doses should be >54 Gy to be considered curative intent.
Loehrer et al. [72] reported the landmark prospective study on sequential radiotherapy for thymomas in 1995. The trial enrolled 23 patients who received induction chemotherapy with the cyclophophamide, doxorubicin and cisplatin regimen and were subsequently treated with definite radiotherapy to a total dose of 54 Gy. This strategy resulted in an overall response rate of 70% and a 5-yr survival of 53%, comparing favourably with the results of incomplete resection [72]. Another prospective trial recruited 25 patients evaluated for radiotherapy as a definite treatment for advanced thymoma [73]. Mean total dose was actually only 46 Gy. Disease-free survival was 81% and overall survival was 72%. Retrospective data in an unselected population reported less impressive results, with disease-free survival of 45–50%.
Definite concurrent chemoradiotherapy with platin and etoposide-based chemotherapy has mainly been used for thymic carcinoma tumours, mostly in a limited subset of patients included in retrospective studies; any recommendation on this strategy is hard to interpret [4].
CHEMOTHERAPY PRINCIPLES
Chemotherapy may be delivered as the sole treatment modality in unresectable, advanced, metastatic or recurrent tumours. Chemotherapy is then a palliative-intent treatment. The aim is to improve tumour-related symptoms by obtaining a tumour response; no prolonged survival is expected. There is currently no rationale to use post-operative or consolidation chemotherapy after R0–R1 resection of a thymoma [6, 22, 55, 74, 75]. Chemotherapy may be delivered in combination with radiotherapy after a R2 resection, as described above. On the contrary, thymic carcinomas do present with frequent and early locoregional and systemic recurrences after surgery [4]. In the landmark Japanese series, 186 patients with thymic carcinoma were included, 92 of which had been completely resected [6]. Outcome was better in patients who received post-operative chemotherapy (5-yr survival 82%, n=12) than in those who received post-operative chemoradiation (5-yr survival 47%, n=24), radiotherapy (5-yr survival 74%, n=33) or no adjuvant treatment (5-yr survival 72%, n=16). Despite the small number of patients, post-operative chemotherapy may then be discussed for thymic carcinoma, usually combined with radiotherapy, especially if no pre-operative treatment was administered [17, 76, 77].
Curative-intent, primary chemotherapy
As stated previously, the ultimate aim of the treatment of invasive thymic epithelial tumours, including stage III–IV tumours that are unresectable at the time of diagnosis, is to achieve complete resection to achieve long-term survival. Several chemotherapy regimens have been used in this setting, mostly consisting of adriamycin- and/or platin-based multi-agent combinations (table 4, fig. 2) [78–88]. Usually two to four cycles of chemotherapy are administered before imaging reassessment. Besides CT scan assessment, PET scan standard uptake value decrease as compared to pre-treatment imaging, which may represent a predictor for resectability [28]. Response rates to chemotherapy ranged from 70% to 80% in the largest studies. Patients for whom R0 resection was thought to be feasible undergo surgery, and complete resection is achieved in approximately 50% of cases (table 4). Post-operative radiotherapy is then delivered frequently. When the patient is not deemed to be a surgical candidate, either because R0 resection is not thought to be achievable, because of poor performance status or a co-existent medical condition, definite radiotherapy, as reported previously, is delivered. As the aim is to increasing the response rate to primary treatment and, thus complete resection rate, chemotherapy may be combined with radiotherapy. Available retrospective data do not provide interpretable data to allow a comparison between chemotherapy and chemoradiotherapy in the pre-operative setting (table 4) [37, 72]. As mentioned previously, a randomised phase II trial evaluating cisplatin and etoposide chemotherapy combined with conformal or intensity-modulated radiotherapy as primary treatment of unresectable thymoma and thymic carcinoma is currently ongoing (Clinicaltrials.gov NCT00387868).
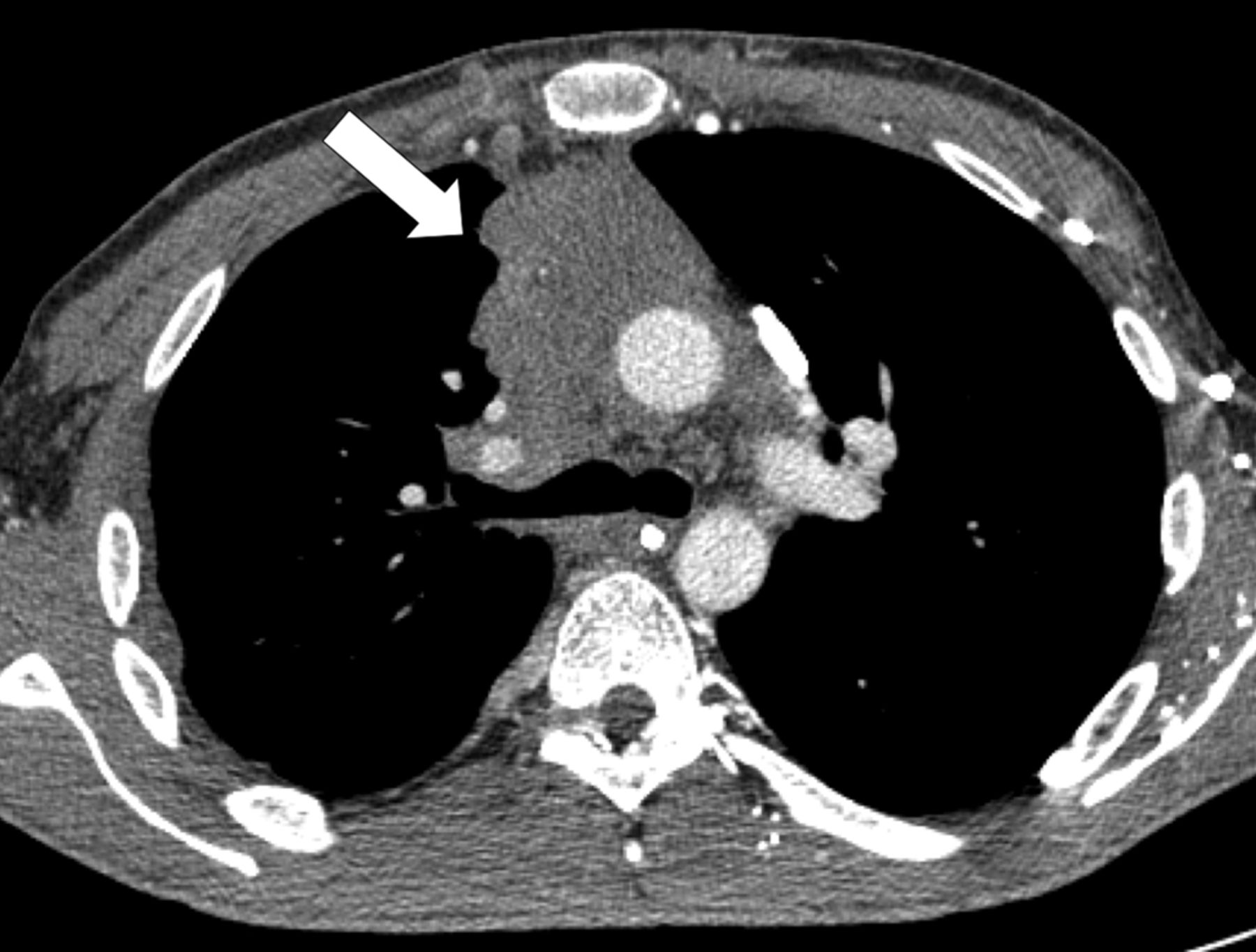
Computed tomography scan of a 37-yr-old male who presented with dyspnoea and fatigue. Invasive tumour of the anterior mediastin was observed in the right hemithorax (arrow); percutaneous biopsy showed a type B3 thymoma. The patient received multimodal treatment with primary chemotherapy followed by surgical resection. Resection status was R1. Post-operative radiotherapy was delivered.
After primary chemotherapy, if radiotherapy is not feasible, either because of a large tumour burden that precludes safe delivery of appropriate doses or because of co-morbidities increasing the risks of radiation-induced toxicity, chemotherapy alone is initiated, in a strategy that may ultimately be considered palliative. In the reported literature, 10–21% of patients with locally advanced thymic tumours receiving upfront chemotherapy did not receive either surgery or radiation therapy or other local treatment. Survival of these patients is frequently limited (table 4).
Overall, the major challenge in interpreting data about pre-operative chemotherapy in thymic malignancies is the wide variation in the number of patients subsequently treated with surgery, radiotherapy or chemotherapy alone, which suggests significant heterogeneity in the inclusion criteria among studies. Response has been evaluated based on elusive criteria in some studies published before CT scans were largely available. In most studies, thymomas and thymic carcinomas, as well as newly diagnosed and recurrent tumours, were not analysed separately. In addition, some investigators distinguished “marginally resectable”, “potentially resectable” and “unresectable” tumours with no clear-cut definition, instead of indicating tumour stage. Ultimately, the majority of studies are retrospective with an uncontrolled design. Finally, one should consider the potential effect of corticosteroids, which have been known for a long time to have a “lympholytic” effect [89]. The thymuses of patients receiving corticosteroids have significantly increased fat and connective tissue, decreased germinal centres and poorer corticomedullary differentiation than those of patients not receiving steroids. In lymphocytic thymomas (type AB, B1 and B2), corticosteroids may then produce a significant reduction of lesion size in imaging studies through lymphocytic depletion, with no anti-tumour effect. Ultimately, primary chemotherapy may provide a window of opportunity to evaluate tumour chemosensitivity before deciding whether surgery or definite radiotherapy may be performed.
Palliative-intent, definite chemotherapy
Palliative chemotherapy is given as the sole treatment modality for thymic tumours, usually in the setting of stage IV, unresectable, recurrent disease. Prolonged disease control is possible, but tumour eradication is not expected. Several studies, both prospective and retrospective, described several regimens for definite chemotherapy (table 5) [90–99], but because there are no randomised studies it is unclear which are best; multi-agent combination regimens and anthracycline-based regimens appear to have improved response rates compared to others, especially the etoposide, ifosfamide and cisplatin combinations. In general, a combination regimen is recommended for at least three and no more than six cycles (fig. 3). Overall, response rates are 20–40% lower than those observed in the preoperative setting. Progression-free and overall survival of patients ranges from 12 to 66 months and from 37 to 72 months, respectively; such variability may be related to the various settings in which chemotherapy was delivered in those studies.

{kind=link}
{kind=link}
{kind=link}
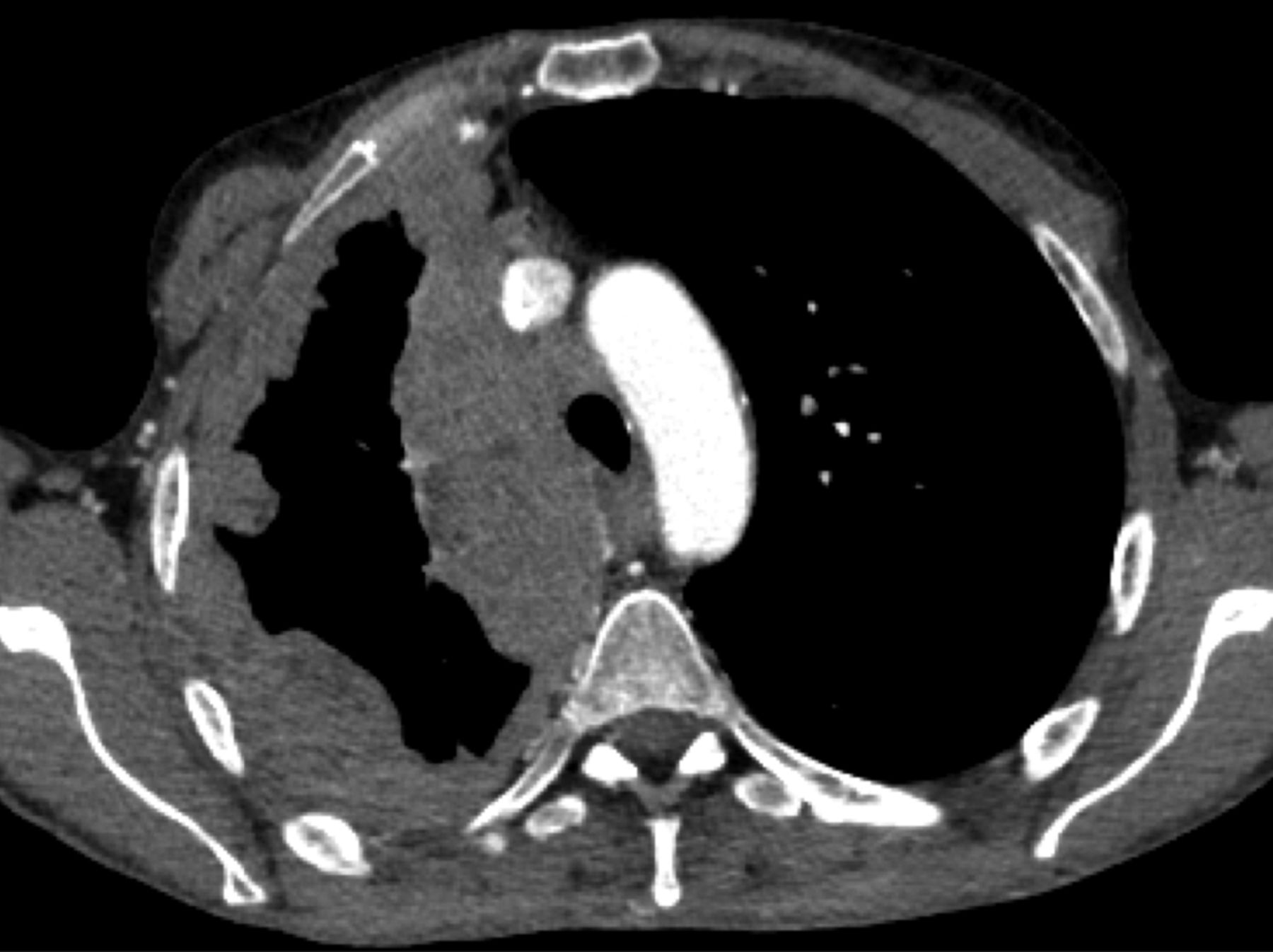
Computed tomography scan of a 45-yr-old female who presented with shortness of breath and chest pain. “Pseudo-mesotheliomatous” pleural invasion was observed in the right hemithorax; surgical biopsy showed a type B1 thymoma. The patient is currently receiving definite chemotherapy with cyclophosphamide, adriamycin, and cisplatin.
In the palliative-intent setting, several consecutive lines of chemotherapy may be administered when the patient presents with tumour progression. It is estimated that 50–70% of patients with thymoma recurrence are eligible for chemotherapy [15]. The strategy may consist of the re-administration of a previously effective regimen [100], especially in case of previous response, late occurring recurrence and, for anthracyclins, a patient in a good medical condition and not having received cumulative doses precluding the safe delivery of at least three additional cycles. The risk of cardiac toxicity is even more increased because of previous use of mediastinal radiotherapy and the potential development of paraneoplastic myocarditis [101]; the use of less toxic agents, including paclitaxel [97] or pemetrexed [92]. In case of recurrence, the strategy may actually primarily consist of a similar multimodal management to that conducted at time of first diagnosis, with surgery and radiotherapy in eligible cases [102–105]. Complete resection remains a major prognostic factor in this setting.
In patients not eligible to receive additional chemotherapy, octreotide may represent a valuable option. As a single agent, octreotide produced objective tumour responses rates ranging from 10% to 37% in tumours showing increased uptake at octreoscan scintigraphy [106–108]. Collectively, 90% of patients with thymoma and 50% of patients with thymic carcinoma experienced disease control, although objective response was reported in only one patient with thymic carcinoma.
TARGETED THERAPIES
Despite the rarity of these tumours, significant efforts have been made to dissect the molecular pathways involved in the carcinogenesis of thymic malignancies [26, 109, 110]. Given the currently available targeted agents outside of a clinical trial, the signalling pathways that are relevant in the clinical care of patients are the KIT and the vascular endothelial growth factor receptor (VEGFR) pathways.
KIT is a transmembrane growth factor with tyrosine kinase activity. Several tyrosine kinase inhibitors targeting KIT have been developed, including imatinib (Novartis, Basel, Switzerland), sunitinib (Pfizer, New York, NY, USA) and sorafenib (Bayer, West Haven, CT, USA), most of which also potently inhibit other kinases, including VEGFR and platelet-derived growth factor receptors. A recent pooled analysis of data reported in the literature indicates that collectively, KIT is overexpressed in 2% of thymomas and 79% of thymic carcinomas [22]. Given the significantly highest frequency of KIT expression in thymic carcinoma, some authors proposed KIT as a diagnostic marker of carcinoma of thymic origin versus other origins in the setting of a mediastinal tumour [111]. Despite the high expression of KIT, KIT gene mutations are found in only 9% of thymic carcinomas (13 out of 128 collectively analysed) (table 6) [26, 112–117]. Clinically, KIT mutant thymic carcinomas then represent a small molecular subset of thymic tumours. The relevance of KIT mutations is even more limited in thymic carcinoma than in other cancers such as gastro-intestinal stromal tumour, as: 1) KIT mutations are far less frequent; 2) KIT expression does not correlate with the presence of KIT mutation; and 3) non pre-treated KIT mutants are not uniformly sensitive to imatinib, based on the clinical and/or the pre-clinical evidence in thymic carcinoma and/or other KIT-mutant tumours (table 6). These findings may explain why the two reported phase II trials with imatinib where patients were not selected [118], or were selected based upon histological type (B3 thymomas and thymic carcinomas) [119] or KIT staining by immunohistochemistry [120] and not upon KIT genotyping were negative.
Multi-kinase inhibitors may also be of interest to target angiogenesis. The most potent pro-angiogenic molecules are those of the VEGF/VEGFR signalling pathway. VEGF-A and VEGFR-1 and -2 are overexpressed in thymomas and thymic carcinomas [121, 122]. Micro-vessel density and VEGF expression levels have been shown to correlate with tumour invasion, aggressive histology and clinical stage [123].
In a phase II trial, bevacizumab was tested in combination with erlotinib in 11 thymomas and seven thymic carcinomas [124]. No tumour response was observed. In a phase I study combining docetaxel with aflibercept, a soluble receptor that binds VEGF-A (also called VEGF trap), one patient with thymoma experienced partial response [125]. Interestingly, despite the large tumour burden of thymic tumours and the frequent abutment to mediastinal vascular structures, no haemorrhagic side-effect has been reported with the use of these drugs in these studies. Beyond the inhibition of KIT, sunitinib and sorafenib also inhibit VEGFR-1, VEGFR-2 and VEGFR-3 at the nano-molar range. The effect of these drugs, especially in KIT wild-type thymic carcinoma tumours may then be partially related to an anti-angiogenic effect [126, 127]. As with sunitinib and sorafenib, motesanib diphosphonate (AMG-706; Amgen, Thousand Oaks, CA, USA) is a specific inhibitor of VEGFR-1, VEGFR-2 and VEGFR-3, which has been reported to control the growth of a thymic carcinoma tumour refractory to chemotherapy for 12 months [128].
Among others, promising targets in thymoma and thymic carcinoma include IGF-1R and histone deacetylase [109, 129]. Cituxumumab, an IGF1-R directed monoclonal antibody was recently reported to produce a promising 90% disease control rate in refractory thymomas [130]. Belisnostat, a histone deacetylase inhibitor, was evaluated in thymic malignancies in a recently completed phase II trial enrolling 41 patients (25 thymomas and 16 thymic carcinomas) [129]. Response and 2-yr survival rates were 8% and 77%, respectively, in thymomas, and 0% and 0%, respectively, in thymic carcinomas.
Given the rarity of the tumour, translation of pre-clinical findings to the clinic may be quick; for example, implementation of KIT genotyping in clinical practice would represent a model of n-of-one trial approach in the field.
The management of thymic tumours is a paradigm of co-operation between clinicians, surgeons and pathologists from establishing the diagnosis to organising the therapeutic strategy and evaluating the prognosis. As a consequence of their rarity, collaborative studies are warranted in order to evaluate and improve current therapeutic standards, taking into account recent advances in techniques and concepts, such as robotic surgery, radiotherapy, molecular biology and supportive treatment. Along with the large variety of questions relative to the treatment strategy, thymic epithelial tumours represent a model of therapeutic implementation and achievement in orphan thoracic oncology, showing how the advent of new results induces new questions, as well as diversifies further clinical research directions and international collaborative initiatives.
Footnotes
Provenance
Submitted article, peer reviewed.
Statement of Interest
None declared.
- Received December 3, 2012.
- Accepted December 18, 2012.
- ©ERS 2013
REFERENCES









