





Systemic sclerosis (SSc) is a systemic autoimmune disease that is characterised by endothelial dysfunction resulting in a small-vessel vasculopathy, fibroblast dysfunction with resultant excessive collagen production and fibrosis, and immunological abnormalities. The classification of SSc is subdivided based on the extent of skin involvement into diffuse cutaneous sclerosis (dcSSc), limited cutaneous sclerosis (lcSSc) or SSc sine scleroderma [1]. While virtually any organ system may be involved in the disease process, fibrotic and vascular pulmonary manifestations of SSc, including interstitial lung disease (ILD) and pulmonary hypertension (PH), are the leading cause of death. As new therapies targeting these pulmonary conditions emerge, early recognition of lung involvement is essential for the care of these patients. In this article we review the direct and indirect pulmonary manifestations of SSc and recent therapeutic trials that have attempted to target these manifestations.
TYPES OF PULMONARY INVOLVEMENT
When a patient with SSc disease presents with signs or symptoms referring to the chest, a number of potential disorders must be considered (table 1) for: direct pulmonary involvement (ILD with or without PH or pulmonary arterial hypertension (PAH), airways disease and pleural involvement); indirect pulmonary complications (aspiration, infection, drug toxicity, malignancy, respiratory muscle weakness, restrictive lung disease from chest wall involvement and lung disease secondary to cardiac involvement); combinations of direct and indirect pulmonary manifestations; and other lung diseases not related to SSc (chronic obstructive pulmonary disease/emphysema, asthma and lung nodules).
DIRECT PULMONARY INVOLVEMENT
In scleroderma, the two most common types of direct pulmonary involvement are ILD and PH, which together account for 60% of SSc-related deaths [2]. While certain pulmonary manifestations may occur more commonly in a subset of SSc (i.e. ILD is more common in dcSSc while PH is more common in lcSSc) [3], all of the known pulmonary manifestations reported have been described in each of the subsets of disease [4]. Pulmonary disease can even occur in SSc with no skin involvement (an entity known as scleroderma sine scleroderma) [5]. These patients can be misclassified as having idiopathic ILD and the presence of telangiectasias, Raynaud’s phenomena, reflux or pericardial effusions; a nucleolar-antinuclear antibody test should alert the clinician to the possibility of scleroderma sine scleroderma [6, 7].
Interstitial lung disease
ILD is common in scleroderma. In early autopsy studies, up to 100% of patients were found to have parenchymal involvement [8, 9]. As many as 90% of patients will have interstitial abnormalities on high-resolution computed tomography (HRCT) [10] and 40–75% will have changes in pulmonary function tests (PFTs) [11, 12]. Parenchymal lung involvement often appears early after the diagnosis of SSc, with 25% of patients developing clinically significant lung disease within 3 yrs as defined by physiological, radiographic or bronchoalveolar lavage (BAL) abnormalities [13]. Risk factors for its development include African–American ethnicity, skin score, serum creatinine and creatine phosphokinase levels, hypothyroidism and cardiac involvement [13, 14]. Genetic factors [15], specific serological findings (anti-topoisomerase [14, 16] and anti-endothelial cell [17] antibodies predict the presence of lung involvement, and anti-centromere and anti-RNA polymerase III antibodies are less associated with lung disease [13, 16, 18]) and the pattern of skin disease (patients with dcSSc have a higher incidence of interstitial disease [3, 19, 20]) all contribute. Predictors of severe restrictive lung disease (defined by a forced vital capacity (FVC) ≤50% predicted) include African–American ethnicity [11], male sex, the degree of physiological abnormalities at diagnosis (FVC and diffusing capacity of the lung for carbon monoxide (DL,CO)) and younger age [11, 21].
Pathogenesis
The pathogenesis of SSc-ILD is not well understood. It is presumed to be related to abnormal interactions between endothelial cells, lymphocytes/monocytes and fibroblasts leading to an excess production of extracellular matrix by fibroblasts in the setting of tissue hypoxia and vascular hyperreactivity [22]. Patients have increased levels of the pro-inflammatory cytokines interleukin (IL)-8, tumour necrosis factor-α and macrophage inflammatory protein-1α in BAL fluid [23]. B-cells may also be involved as patients with SSc-ILD have higher levels of anti-topoisomerase antibodies [24] and anti-fibroblast antibodies [25], the latter having been shown to activate fibroblasts and induce extracellular matrix production [26].
Radiology
HRCT is the standard method for the noninvasive diagnosis of SSc-ILD and can detect mild abnormalities. The true incidence of HRCT abnormalities is difficult to determine, but the majority of patients (55–84%) will have disease [16, 19, 27–29] and the extent is generally limited with an average of 13% of the parenchymal involved [28, 30]. Despite the sensitivity of HRCT in SSc-ILD there are limitations. It can be normal in patients with PFT abnormalities, and a number of those with an abnormal chest examination (i.e. crackles) develop abnormal HRCT scans at follow-up [19]. In spite of these limitations, the presence of a normal HRCT at baseline predicts a low likelihood for the development of SSc-ILD, as 85% of these patients still have a normal HRCT at a mean follow-up of 5 yrs [19].
The HRCT pattern seen in SSc patients is generally nonspecific interstitial pneumonia (NSIP) [30], with a greater proportion of ground-glass opacities (GGOs) and a lower degree of coarse reticulation (fig. 1). However, a usual interstitial pneumonia (UIP) pattern can also be seen (fig. 2). Honeycomb cysts can be seen in up to a third of patients with SSc-ILD and are more common in patients with lcSSc [31]. The pattern seen on HRCT predicts the underlying histopathology, with reticulation representing underlying fibrosis on biopsy and consolidation representing inflammation [32]. Reversibility of HRCT changes is rare [29]. Instead, the radiographic progression seems to be one of replacement of GGOs with honeycombing/traction bronchiectasis and/or bronchiolectasis over time [19]. Up to two-thirds of patients with GGOs progress to fibrosis, regardless of therapy [33].

High-resolution computed tomography from a patient with systemic sclerosis showing basilar predominate reticulation and ground-glass opacities with an absence of significant honeycombing in a pattern consistent with nonspecific interstitial pneumonia. The patient also has an air–fluid level in the oesophagus consistent with scleroderma-associated oesophageal dysfunction.

High-resolution computed tomography from a patient with systemic sclerosis showing peripheral and basilar predominate reticulation and honeycombing with an absence of significant ground-glass opacities in a pattern consistent with usual interstitial pneumonia. The patient also has an air-filled oesophagus consistent with scleroderma-associated oesophageal dysfunction.
Pulmonary function tests
Screening pulmonary physiology shows a reduction in FVC in 40–75% of patients, with 15% having a severe reduction [11, 12, 34]. DL,CO is reduced in almost all patients with other PFT abnormalities [35] and correlates with the extent of lung disease on HRCT [36]. DL,CO levels (corrected for haemoglobin) are lower in patients with UIP on biopsy [35] and, although FVC and DL,CO are both identified as adverse prognostic markers [11, 21], a declining DL,CO is the single most significant marker of poor outcome [35].
BAL cellular profile
BAL fluid from healthy never-smokers contains a predominance of macrophages (80–90%) with lower percentages of lymphocytes (5–15%) and neutrophils (≤3%) [37]. An abnormal BAL cellular profile (defined as a neutrophil count ≥3% or an eosinophil count ≥2% on BAL) is present in 38–72% of SSc patients with parenchymal involvement on HRCT [38], but 50% of patients with a normal HRCT will also have abnormal BAL cell counts [33]. Early studies suggested that patients with an abnormal BAL who did not receive immunomodulatory therapy had a progressive decline in FVC and DL,CO when compared to those with a normal BAL [39, 40]. However, subsequent studies determined that this prior association between BAL cellularity and disease progression may have been an epiphenomena; these data do not add to the prognostic evaluation when PFTs and HRCT are available [41, 42]. Sampling error may play a significant role in explaining some of these discordances [43]. The current use of BAL cellular analysis for SSc lung disease is limited to excluding infection and for research purposes.
Pathology
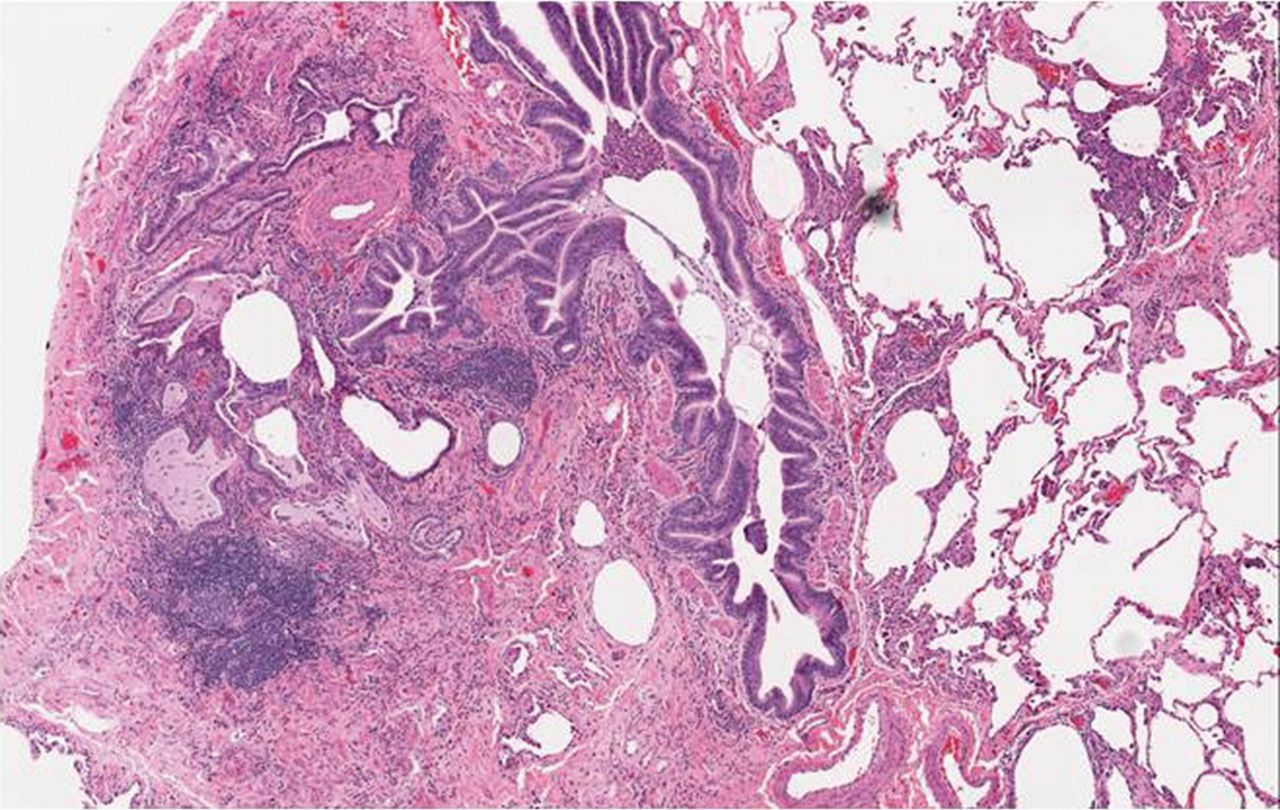
On surgical lung biopsy, a mixed pattern of fibrosis and inflammation is seen in the majority of cases. After Katzenstein and Fiorelli [44] described the features of NSIP in 1994, a re-evaluation of patients with SSc-ILD revealed a significant number of patients with this pattern (fig. 3) [45]. In the largest study to date, 77% of patients with SSc-ILD had a histological pattern of NSIP, the majority of which were fibrotic NSIP [35]. A UIP pattern can occasionally be seen and, when compared to those with IPF, more germinal centres and inflammation and less fibroblastic foci are noted (fig. 4) [46].

Histopathology results from a patient with systemic sclerosis and nonspecific interstitial pneumonia showing cellular interstitial infiltrates in a temporally uniform distribution.

Histopathology results from a patient with systemic sclerosis and usual interstitial pneumonia showing patchy interstitial fibrosis in close proximity to unaffected lung tissue.
Treatment
Agents such as corticosteroids have historically been used, but their efficacy has never been proven in SSc-ILD. Two retrospective studies found an association with scleroderma renal crisis when higher doses were used in patients with dcSSc [47, 48]. If used, doses of ≤15 mg·day−1 are generally recommended. d-penicillamine has been used, with a retrospective analysis showing that it led to improvements in DL,CO [49]. Its use is limited by adverse effects [50], and no prospective trials looking at its effect on SSc-ILD have been conducted. A prospective trial of interferon-γ found no significant effect in SSc-ILD [51]. Mycophenolate mofetil (MMF) has been used with increasing frequency and has a good safety profile [52]. In small, retrospective series, improvement in skin score, stability (if not improvement) in PFTs, and improved survival have all been reported [53–57]. Small open-label trials with MMF either following anti-thymocyte globulin [58] or used in conjunction with low-dose prednisone [59] showed stable or improved PFTs and HRCT findings. Mycophenolate was well tolerated in patients with diffuse SSc and ILD and associated with lower rate of decline in FVC and survival compared to other immunosuppressive agents [60].
There are more robust prospective data with cyclophosphamide (CYC). Early studies with CYC, dating back to 1993, showed that SSc-ILD patients treated with CYC and prednisone had significant improvement in FVC at 6 and 12 months [61–63]. In 2000, a retrospective cohort study found that patients with abnormal BAL findings who were treated with CYC were more likely to have stabilisation or improvement in FVC and DL,CO than those who were not treated [40]. This data led to two prospective, randomised, placebo controlled trials of CYC in SSc-ILD. The first, the Scleroderma Lung Study (SLS), was a 13 centre double-blind placebo-controlled trial looking at 1 yr of oral CYC in patients with active symptomatic scleroderma lung disease [64]. Results showed a small but significant positive treatment effect on FVC as well as improvements in dyspnoea and skin thickness. More adverse events were noted in the CYC group but no significant increase in serious adverse events. The greatest benefit was seen in subjects with more fibrotic lung disease on HRCT [65]. When patients were evaluated 1 yr after stopping therapy (24 months into the trial), benefit accrued to 18 months and then waned to placebo levels with the exception of the improvement in dyspnoea [66].
A second trial called the Fibrosing Alveolitis in Scleroderma Trial looked at intravenous CYC for 6 months followed by azathioprine [67]. No statistical differences were found in FVC, DL,CO, HRCT appearance or dyspnoea scores (although there was a trend towards a significant improvement in FVC) [67]. A recent meta-analysis of the effects of CYC on pulmonary function found no significant improvement with treatment [68]. Due to the preliminary data with MMF and the lack of sustained improvement seen with CYC, the SLS-II is currently looking at 1 yr of CYC versus 2 yrs of MMF in the treatment of SSc-ILD.
Other agents have been evaluated in smaller trials. The tyrosine kinase inhibitor imatinib [69], anti-CD20 therapy with rituximab [70, 71] and anti-CD25 therapy with basiliximab [72] have been tested in small open-label trials and there are plans for additional agents to be tested in prospective, randomised controlled trials.
Haematopoietic stem-cell transplant (HSCT) has been evaluated for its possible role in treating the immune activation in SSc. Early non-randomised studies showed decreases in HRCT disease extent scores and improvement in oxygenation [73, 74]. A follow-up open-label randomised phase-II trial looking at HSCT compared with pulse CYC (the ASSIST (Autologous Non-Myeloablative Haemopoietic Stem-Cell Transplantation Compared With Pulse Cyclophosphamide Once Per Month For Systemic Sclerosis) trial) found improvements in lung function and HRCT scans up to 2 yrs after transplantation [75]. There are ongoing open-label trials actively recruiting to further investigate the role of stem-cell transplant in the management of SSc. In the USA, the ongoing SLS-II is comparing the efficacy of treatment with mycophenolate with CYC. Thus, it is hoped that the efficacy of treatment for SSC-ILD with these treatment interventions will be ascertained in the near future.
Outcomes
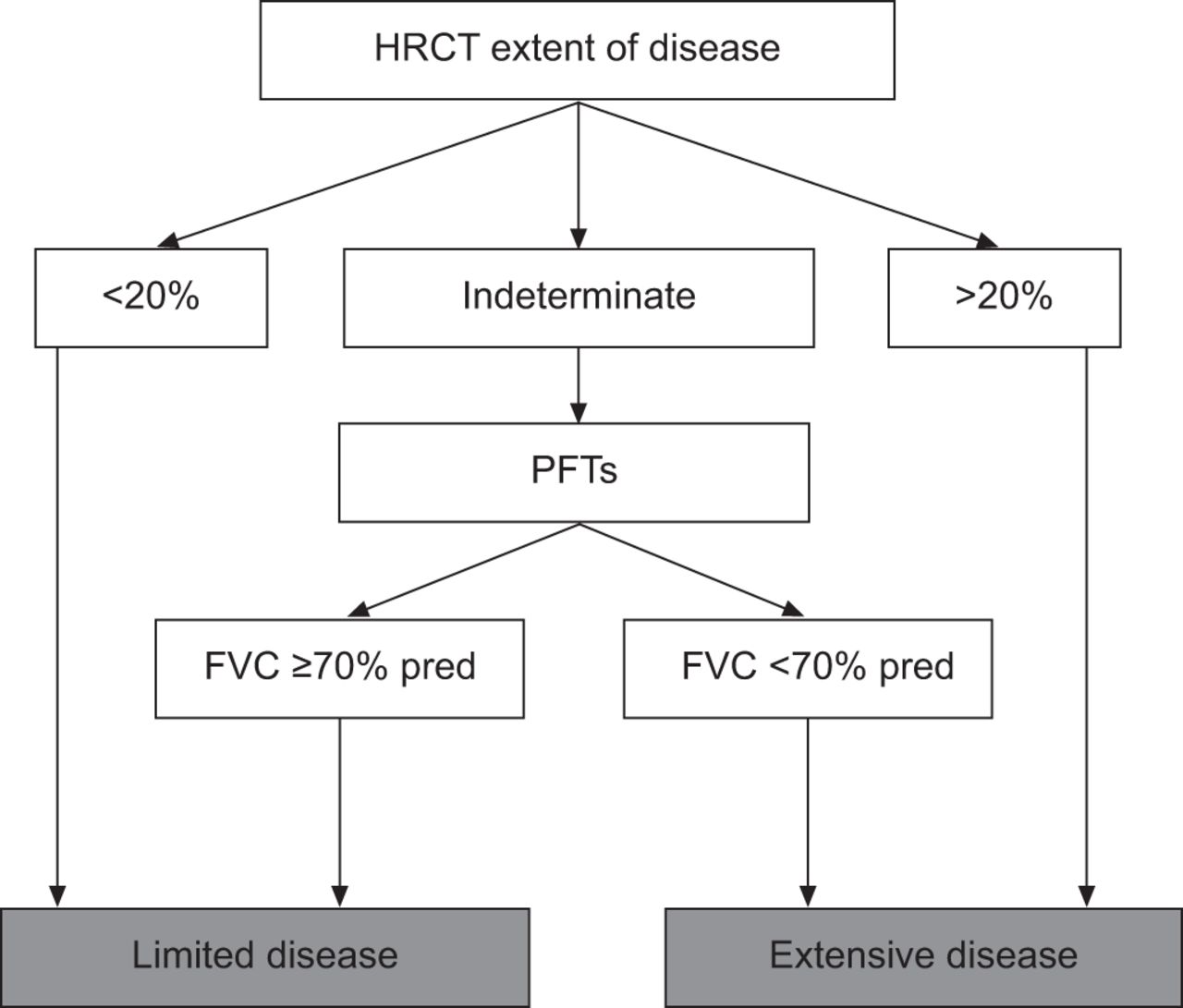
Early death from SSc-ILD is relatively uncommon with an estimated survival of 85% at 5 yrs [76]. Severe restrictive lung disease (defined by an FVC ≤50% pred) has been reported to occur in 13% of patients [11]. Patients who develop severe ILD tend to have progressive decline in lung function within the first 2 yrs of disease [11]. Unlike the idiopathic interstitial pneumonias, survival does not appear to differ between those with a pathological pattern of NSIP and those with UIP [35]. Both histological groups have an 82–90% 5-yr survival and 29–69% a 10-yr survival [35]. It also appears that the subtype of scleroderma (limited versus diffuse) does not affect the likelihood of progression [31]. When followed over time, reductions in DL,CO at 3 yrs and increased eosinophils on BAL were associated with a decreased survival [35]. Recently, Goh et al. [28] developed a prognostic algorithm for patients with SS-ILD. The algorithm relies solely on HRCT scoring for mild or severe cases with recourse to a FVC cut-off in cases of indeterminate extent of disease. This staging system was shown to be easy to use and predictive of mortality (fig. 3).
Pulmonary hypertension
PH can occur in all forms of SSc and is associated with early mortality. Along with mixed connective tissue disease, patients with SSc have the highest prevalence of PH among patients with a collagen vascular disease (CVD) [77]. The updated clinical classification of PH divides patients into five groups based on the aetiology of their PH [78]. SSc patients may fall into group 1 (isolated PAH, defined as a resting mean pulmonary artery pressure (mPAP) >25 mmHg with a pulmonary capillary wedge pressure ≤15 mmHg [79]), group 2 (PH resulting from left ventricular involvement or diastolic dysfunction) and group 3 (PH resulting from ILD/hypoxaemia). Confounding this issue, patients can have combinations of these various forms of PH.
The prevalence of PAH in SSc (SSc-PAH) is variable and depends on the method of detection and the population studied. Using transthoracic Doppler echocardiography to screen SSc patients, the prevalence of PAH has been reported to range from 13% to 35% [80, 81]. However, when right heart catheterisation (RHC) is performed on “high-risk” SSc patients (defined as a combination of abnormal echocardiography findings, reduced DL,CO in the absence of pulmonary fibrosis, a precipitous fall in DL,CO and/or unexplained dyspnoea), a prevalence of 7–13% is noted [82–85]. PAH can develop anytime during the course of SSc [86] and is more common in lcSSc when compared to diffuse disease [87, 88]. In the European League Against Rheumatism (EULAR) Scleroderma Trials and Research database, a multinational open scleroderma cohort with over 3,000 patients, isolated PAH was seen in 9.2% of lcSSc and 5.8% of dcSSc patients. The South Australian Scleroderma Register, a population-based registry with 608 patients, found PAH in 11% of patients with scleroderma; all of whom had lcSSc [89].
Multiple risk factors have been identified including increased age at diagnosis [90], more severe Raynaud’s phenomena [91], the presence/severity of digital tip ulcers [89, 91, 92], a diagnosis of lcSSc/CREST (calcinosis, Raynaud’s phenomenon, oesophageal dysmotility, sclerodactyly and telanginectasia) syndrome [93], decreased nailfold capillary density [94] and increased numbers of telangiectasias on examination [89]. Specific autoantibodies, including the presence of anti-U3 ribonucleoprotein antibodies [91, 95], anti-topoisomerase IIα antibodies [96], and anti-centromere antibodies [91, 97], appear to be associated with a higher risk of PAH as do higher erythrocyte sedimentation rates and immunoglobulin G levels [92]. The presence of anti-Scl70 antibodies is associated with progressive ILD and appears to be less associated with PAH [91]. Patients with SSc-PAH are older, more severely ill and more likely to be female when compared to idiopathic PAH (IPAH) [98].
Pathogenesis
The pathogenesis of SSc-PAH is unclear. The pathogenesis seems to be one of injury of the vascular endothelium with subsequent apoptosis, inflammation and dysregulated angiogenesis with subsequent arterial obliteration and narrowing from fibrosis. Genetic studies have revealed that these patients are more likely to have the presence of class I human leukocyte antigen-B35 [96] and an absence of bone morphogenic protein receptor-2 mutations (seen in 25–50% of familial and sporadic IPAH) [99]. Patients with SSc-PAH also have notable cellular and humoral abnormalities. In gene expression analysis, lung tissue from patients with SSc-PAH have an up-regulation in genes involved in antigen presentation, chemokine pathways and metallothionein expression (involved in hypoxia-induced vasoconstriction); patterns similar to those seen in IPAH [100]. Peripheral blood mononuclear gene expression can distinguish SSc patients with PAH from those without PAH. IL-7r and CCR7 were differentially expressed in patients with SSc-PAH [101]. Antibody expression is also altered; antibodies directed against endothelial cell antigens (which target lamin A/C and β-tubulin [102] activate endothelial cells and lead to apoptosis [103, 104]) and fibroblasts (which could activate fibroblasts and induce collagen production [105]) have been reported. Serum biomarkers known to be involved in vascular and endothelial activation have been studied in SSc-PAH. Higher levels of endothelin-1 (a potent vasoconstrictor), IL-8 (a chemokine produced by pulmonary fibroblasts and alveolar macrophages) and endoglin (a glycoprotein expressed by endothelial cells) are seen in patients with SSc-PAH [106]. Growth differentiation factor (GDF)-15 is a cytokine involved in cell growth and differentiation, cell-to-cell signalling and apoptosis regulation [107]. Levels of GDF-15 are elevated in patients with SSc-PAH, correlate with pulmonary artery pressures and have increased expression on the lung tissue of patients with SSc-PAH [108].
Echocardiography
Transthoracic echocardiography is the most widely used tool to screen for PAH in SSc. The performance characteristics of echocardiography depend on the population evaluated and the cut-off used. Studies show that 55–86% of patients with an echocardiography suggestive of PH (right ventricular systolic pressure (RVSP) 30–40 mmHg or higher with or without symptoms) will have PH on RHC [85, 109]. Higher cut-off points for RVSP as well as incorporating other characteristics of increased pulmonary pressures, such as increased right atrial or right ventricular size, decreased right ventricular function or reduced pulmonary artery acceleration times, increases the specificity of echocardiography for the diagnosis of PH. False-positive and -negative results regularly occur and tend to do so in patients with mild disease; false-negative results have been reported in patients earlier in the course of disease [85].
Radiology
Chest radiography is the least sensitive test for PAH but shows the greatest specificity (up to 100% in one study) [88]. Findings include enlargement of the right pulmonary artery (>1.1 cm), loss of peripheral vasculature (“pruning”) and filling of the retrosternal space by the right ventricle on the lateral images [110]. Predictive findings on HRCT are the mean pulmonary artery diameter and the ratio of the mean pulmonary artery diameter to the ascending aorta diameter [111]. Abnormalities in the pericardium, specifically thickening as measured by the total pericardial score, are associated with echocardiographic evidence of PAH [112].
Pulmonary function tests
Reductions in DL,CO are common in SSc; Steen et al. [113] found isolated reductions in DL,CO in 19% of all SSc patients, but only a minority developed PAH. However, a moderate reduction (DL,CO <55% pred) in association with an FVC/DL,CO ratio greater than 1.4% [113] or a DL,CO that is low or declining in the absence of parenchymal lung disease predicts the presence or future development of PAH [89, 111]. Hachulla et al. [83] found that a DL,CO <60% pred in the absence of parenchymal lung disease was significantly associated with PAH (OR 9.23, 95% CI 2.73–31.15). Steen and Medsger [91] found a significantly lower DL,CO in patients with PAH (52% versus 81%, p<0.001) at an average of 4.5 yrs before the diagnosis of PAH and found that a declining DL,CO over 15 yrs strongly predicts the development of PAH. Ungerer et al. [88] found that a DL,CO <43% pred had the greatest sensitivity of any single diagnostic test (67%).
Right heart catheterisation
RHC is the gold standard for diagnosis in SSc-PAH and must be performed prior to initiation of treatment to confirm the diagnosis of PAH, characterise the severity of disease and assist with the selection of appropriate therapy. For example, patients with elevation of right atrial pressures and decreased cardiac output should be considered for intravenous or subcutaneous prostanoid therapy, while those with less severe disease may be considered for oral or inhaled therapy.
Pathology
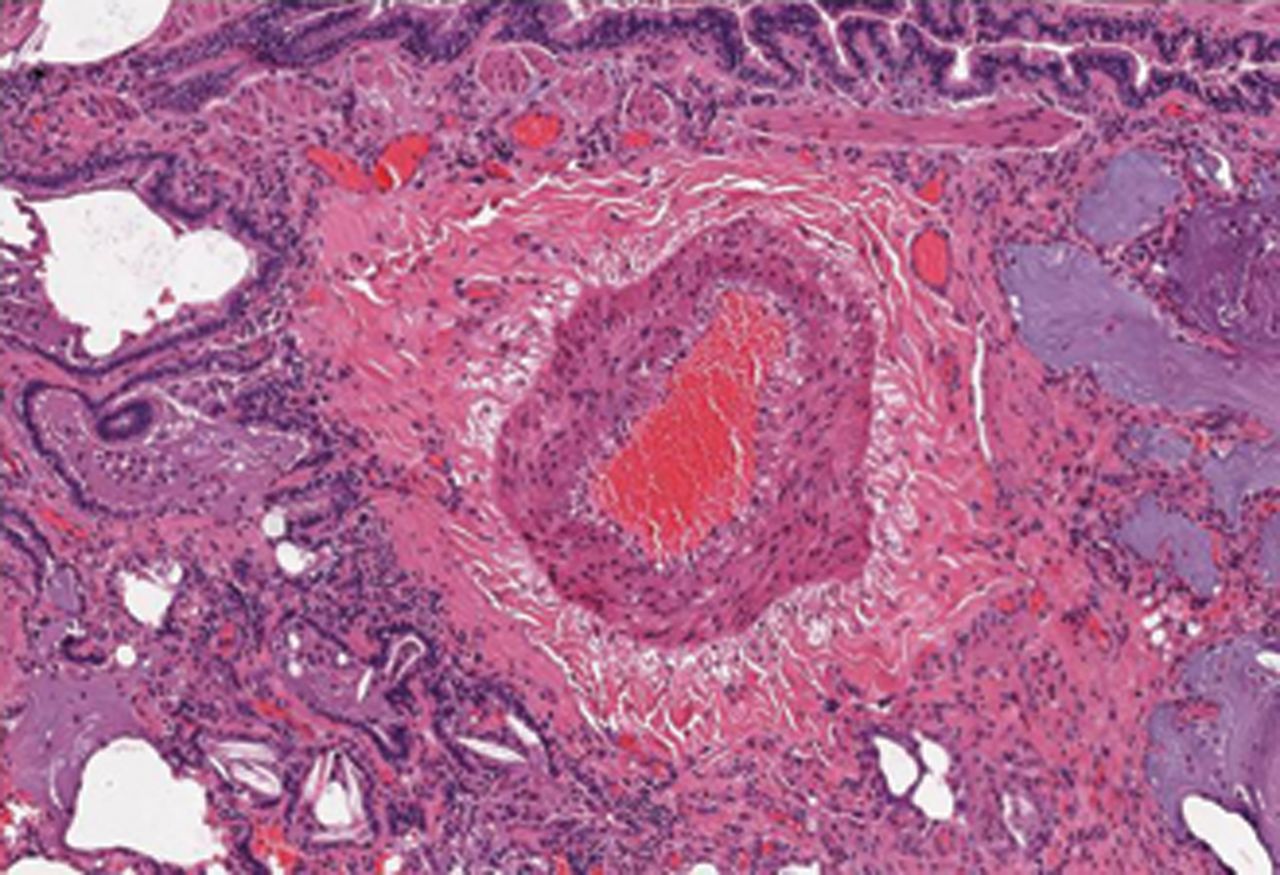
Early autopsy studies found abnormalities in the pulmonary vasculature in nearly 50% of patients with SSc [114], with changes primary involving the small and medium muscular arteries [115]. The primary findings are intimal fibrosis (affecting the small distal vessels adjacent to the alveoli), medial hyperplasia and adventitial fibrosis affecting the pulmonary arterioles (fig. 5). This intimal fibrosis leads to concentric obliterative lesions in pulmonary vessels and luminal occlusion [116, 117]. Intimal fibrosis is also seen in veins and venules and is occasionally associated with a pulmonary veno-occlusive disease (PVOD) pattern [118]. Eccentric intimal fibrosis indicating previous thrombosis is seen in a number of cases [118]. Plexiform lesions, commonly found in IPAH and some forms of secondary PH, have been observed in SSc-PAH by some investigators [117] but not others [118]. The small vessel intimal fibrosis (with involvement of the veins/venules), the presence of a PVOD pattern, the presence of concentric laminar intimal fibrosis and the paucity or absence of plexiform lesions sets SSc-PAH apart from IPAH [118].

A pulmonary arteriole from a patient with systemic sclerosis-associated pulmonary artery hypertension showing significant medial hypertrophy.
Treatment
There are currently a number of medications approved for the treatment of PAH [119]. Medications include calcium-channel blockers (nifedipine, diltiazem and amlodipine), prostacyclin analogues (epoprostenol, iloprost, treprostinil and beraprost), endothelin receptor antagonists (bosentan and ambrisentan) and phosphodiesterase type 5 (PDE5) inhibitors (sildenafil and tadalafil) [120]. While only one study to date has specifically studied the impact of treatment in scleroderma (intravenous epoprostenol in patients with SSc-PAH) [121], many studies have included patients with SSc-PAH. Although none of these studies have documented a survival benefit, many have demonstrated improvement in surrogate end-points, such as exercise tolerance or time to clinical worsening. Again, in the majority of these clinical trials, SSc-PAH comprised a subset of the World Health Organization (WHO) class 1 PAH patients being studied making it difficult to draw specific conclusions regarding the impact of these medications in SSc. Patients with SSc-associated PAH are generally the least responsive to therapy and have a significant mortality [122].
Prostacyclin is produced by the vascular endothelium, has vasodilating and anti-platelet functions and is reduced in adults with PH [123]. Badesch et al. [121] found that the addition of epoprostenol (a naturally occurring prostacyclin analogue) to conventional therapy in patients with moderate-to-severe SSc-PAH (with mPAP ≥35 mmHg) led to improvements in haemodynamics, exercise capacity and symptoms. Treprostinil, a prostacyclin analogue with a longer half-life then epoprostenol, was evaluated in patients with PAH, including patients with CVD, the majority of whom had SSc. The treatment group, which included patients with SSc, had a 25-m improvement in placebo-corrected 6-min walk distance (6MWD), improved haemodynamics (increased cardiac index and reduced pulmonary vascular resistance index) and improved dyspnoea scores [124]. Both inhaled iloprost and inhaled treprostinil have been proven efficacious in the treatment of patients with PAH in other studies that have included patients with SSc-PAH [125–127].
Bosentan is an oral competitive antagonist of endothelin-1 and non-selectively blocks both endothelin receptors A and B (ETA and ETB). Endothelin-1 is an endogenous vasoconstrictor and smooth muscle cell mitogen that is overexpressed in patients with PAH [128]. In a subgroup analysis of the BREATHE (Bosentan Randomised Trial of Endothelial Antagonist Therapy)-1 trial, bosentan had a nonsignificant placebo-corrected improvement in the 6MWD by 43 m in patients with SSc-PAH [129]. This placebo corrected improvement represented stabilisation in 6MWD (an absolute improvement by only 3 m), a contrast to the 46-m absolute improvement in 6MWD seen in patients with IPAH. Girgis et al. [130] reviewed their experience with bosentan in IPAH and SSc-PAH and found that only 25% of their patients with SSc-PAH had a functional class improvement and 47% failed therapy with either lack of clinical effect or hepatotoxicity. The majority of SSc-PAH patients had stabilisation or decline and had a worse survival compared to IPAH (2-yr survival 79% versus 100%) [130]. Finally, an open label study of bosentan in PAH associated with CVD (the majority of whom had SSc) found a WHO functional class improvement in 27%, a 48-week survival of 92% and no significant changes in quality of life [131]. Ambrisentan is a specific ETA antagonist. The AIRES (Ambrisentan in Pulmonary Arterial Hypertension, Randomised, Double-blind, Placebo-Controlled , Multicenter, Efficacy) study demonstrated improved exercise capacity in patients with PAH that included a subset of patients with SSc [132]. Results from the recently completed SERAPHIN (Study with Endothelin Receptor Antagonist in Pulmonary Arterial Hypertension to Improve Clinical Outcome) study show that macitentan led to a reduction in morbidity and mortality compared to placebo [133]. It has been approved for use in Europe and has a new drug application pending in the USA.
Inhibition of PDE5 leads to accumulation of intracellular cyclic guanosine monophosphate, enhancement of nitric oxide-mediated vasodilation and reduction in the proliferation of smooth muscle cells in the pulmonary vasculature. Sildenafil citrate is a selective PDE5 inhibitor and has been evaluated in PAH. A post hoc analysis of the SUPER (Sildenafil Use in Pulmonary Arterial Hypertension) study found that patients in that trial with CVD, most of whom had SSc, had increases in 6MWD (55 m at the lowest dose), decreases in mPAP and pulmonary vascular resistance and an improvement in WHO functional class in 29–42% [134]. Oral tadalafil was also demonstrated to be efficacious in the treatment of PAH in the PHIRST (Pulmonary Arterial Hypertension and Response to Tadalafil) trial. Again, patients with SSc-PAH were included in this study although no separate subgroup analysis was performed in this patient population [135].
Outcome
The presence of PAH in SSc has a major negative impact on survival; it is the second most frequent cause of death behind ILD, causing close to 30% of all deaths [2]. Survival in newly diagnosed patients with SSc-PAH is 49–56% at 3 yrs after diagnosis [84, 93]. Patients with SSc-PAH also have a three-fold increase in mortality when compared to other forms of PAH (sporadic, familial and anorexigen use) [136]. There are other identified factors that increase mortality in these patients, such as high initial pressures and rising pressures [81] and indices of right heart failure such as elevated mean right atrial pressure, raised mPAP and low cardiac index [84]. Patients with CREST and PAH have a 2-yr cumulative survival rate of 40% [87]. Older patients and those with limited disease are more likely to progress to severe PAH [137]. In patients on treatment, unrecognised PVOD may contribute to refractoriness to therapy [118].
Other causes of PH in SSc
Undiagnosed cardiac disease is a potential contributor to elevated pulmonary pressures in SSc (WHO class II PH). Recent techniques such as tissue Doppler echocardiography [138, 139] and cardiac magnetic resonance imaging [140] are able to detect cardiac dysfunction in SSc patients early in disease, even when asymptomatic with normal conventional echocardiogram. Furthermore, evidence exists that in patients with SSc-PAH, right ventricular contractility is reduced out of proportion to the PH [141].
PVOD is a rare, but important, cause of PH in SSc patients. PVOD, characterised by intimal proliferation of the intrapulmonary veins and venules, results in a post-capillary form of PH. Imaging features may include interlobular septal thickening, centrilobular GGO and pleural effusions. It is important to distinguish PVOD from SSc-PAH as therapy with vasodilators may result in respiratory failure [142].
Combined PH and SSc-ILD
Patients with SSc can present with both ILD and PH. This subtype of PH falls into group 3 in the current PH classification [78]. The prevalence of isolated PAH and PH with lung disease by echocardiography seems similar across studies; 18–22% for both groups [143, 144]. Patients with PH and ILD appear to have been diagnosed with SSc at an older age, are older than patients with ILD alone, and have a higher incidence of anti-topoisomerase positivity and dcSSc when compared to SSc-PAH alone [143]. Patients with PH and ILD also appear to have a significantly lower arterial oxygen tension when compared to those with isolated PH [144]. The likelihood of PH increases with more severe restriction; 50% of patients with a FVC <50% have echocardiography evidence of PH [143]. In the setting of concomitant ILD, DL,CO does not correlate with systolic pulmonary artery pressures [144]. A subset of these patients will have PH out of proportion to their lung disease (33% in one series) [144]. It is possible that these patients have other undiagnosed contributors to PH (i.e. chronic thromboembolic disease and untreated sleep apnoea). Patients with combined disease have a mortality risk ratio of 2.4 when compared to SSc alone [143], 3-yr survival rates of 39% compared to 64% in SSc-PAH [143] and a mortality of five-fold greater than SSc-ILD alone [145]. A multivariate analysis revealed a five-fold increase in the risk of death in combined disease compared to SSc-PAH alone [146].
Suggested approach for the clinical management of patients with scleroderma lung disease
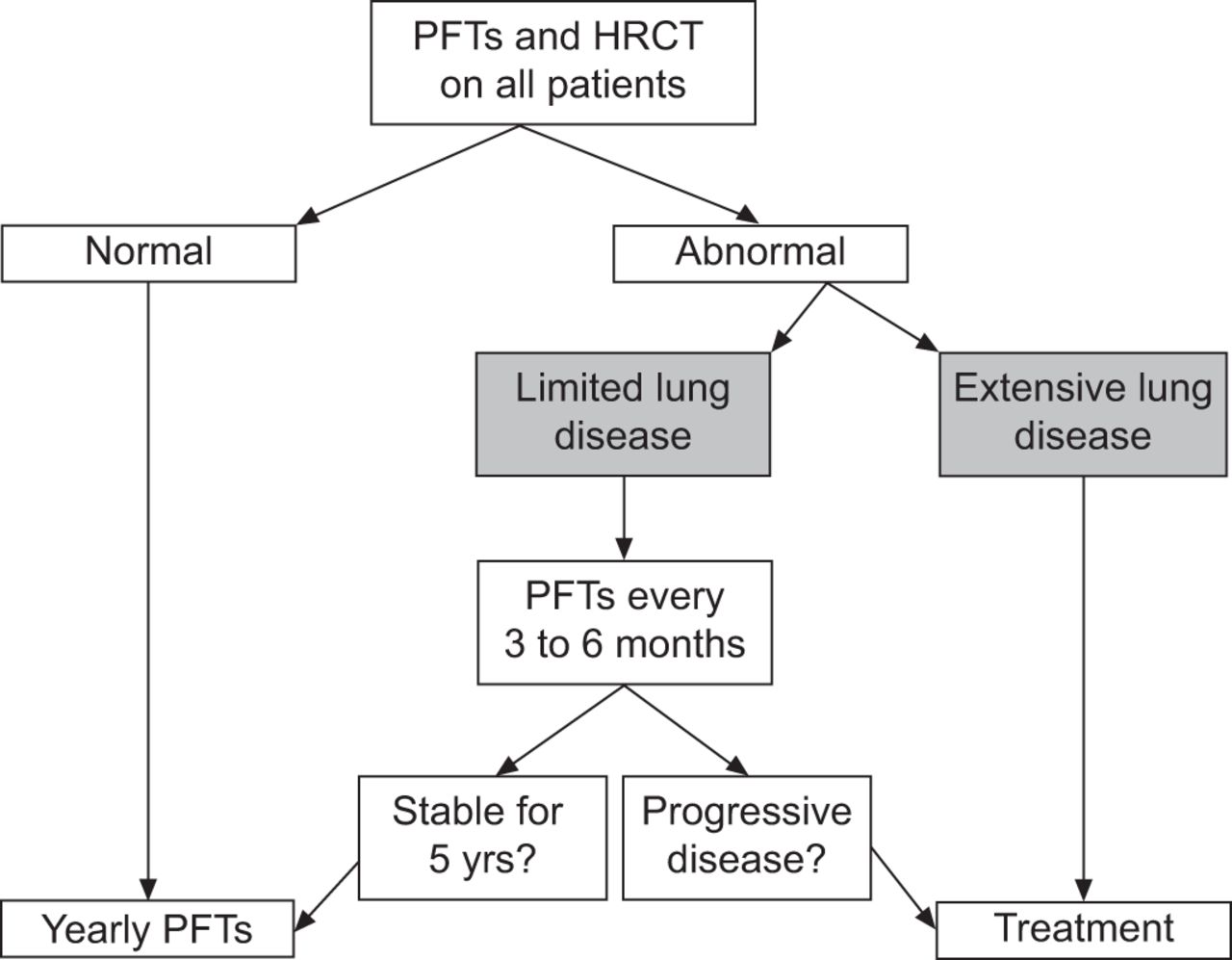
As patients who develop significant and progressive SSc-ILD tend to do so early after the SSc diagnosis, clinicians should consider PFTs and HRCT chest imaging in all patients to facilitate the early identification of those at risk for the development of clinically important ILD. Normal results of this testing portends a good prognosis. Of those with measurable disease, the extent of the abnormalities identified is important as patients with mild physiological or imaging abnormalities are likely to remain clinically stable indefinitely while those with more severe disease are at increased risk for disease progression [28]. A simple stratification scheme developed by Goh et al. [28] utilises HRCT extent of disease and PFTs and provides discriminatory prognostic information (fig. 6). Surgical lung biopsy in these patients is not routinely necessary as the clinical course and outcome is similar between the major histopathological subsets in SSc-ILD (i.e NSIP and UIP) [35]. It should be reserved for atypical imaging presentations and when the diagnosis is unclear. The decision to treat should be individualised and made on the basis of the clinical significance of the disease and the likelihood of future progression (fig. 7). In patients with mild and stable radiographic or physiological derangements, clinicians should consider querying symptoms and physiology every 6 months for the first 5 yrs. After stability is confirmed, less frequent testing seems reasonable. Changes in physiological variables such as declines in FVC or DL,CO should prompt chest imaging and evidence of disease progression should prompt a discussion regarding the appropriateness of treatment. Consideration should be given to CYC for induction and MMF for maintenance therapy in patients requiring treatment.

Extent of interstitial lung disease (ILD) in patients with systemic sclerosis-associated ILD. A simple stratification that utilises pulmonary function tests (PFTs) and extent of disease on high-resolution computed tomography (HRCT) to provide discriminatory prognostic information. FVC: forced vital capacity. Reproduced from [28] with permission from the publisher.

A suggested approach for the long-term follow-up of patients with systemic sclerosis-interstitial lung disease. PFTs: pulmonary function tests; HRCT: high-resolution computed tomography.
As the prevalence of PAH exceeds 10%, its presence increases morbidity and mortality and effective treatments are available [147], screening for PAH is appropriate. Chest symptoms (pain and dyspnoea), signs (lower extremity oedema and pronounced second heart sound), physiological abnormalities (low or reducing DL,CO or FVC/DL,CO >1.4%) and radiographic abnormalities (increase mean pulmonary artery diameter) should prompt earlier or more frequent screening. Vachiery and Coghlan [147] devised an algorithm based on published management guidelines for PAH that recommends yearly echocardiography on all patients and the use of biomarkers with borderline echocardiography findings [148]. Hachulla et al. [83] recommend echocardiography on all SSc patients and using symptoms on borderline echocardiography findings. Sweiss et al. [110] devised a diagnostic algorithm that included an evaluation for other causes of PH (to look for clinical group 2 and 3), as well as recommended treatment options (fig. 8). The ongoing DETECT trial is a prospective cohort study investigating noninvasive screening tools and clinical findings for the ability to predict PAH in SSc patients [149]. Current EULAR guidelines recommend bosentan as first-line therapy with consideration given to use of sildenafil and intravenous epoprostenol [150]. Recent data suggest that patients with early or “borderline” SSc-PAH are likely to have an increase in their mPAP [151] and may benefit from treatment [152].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
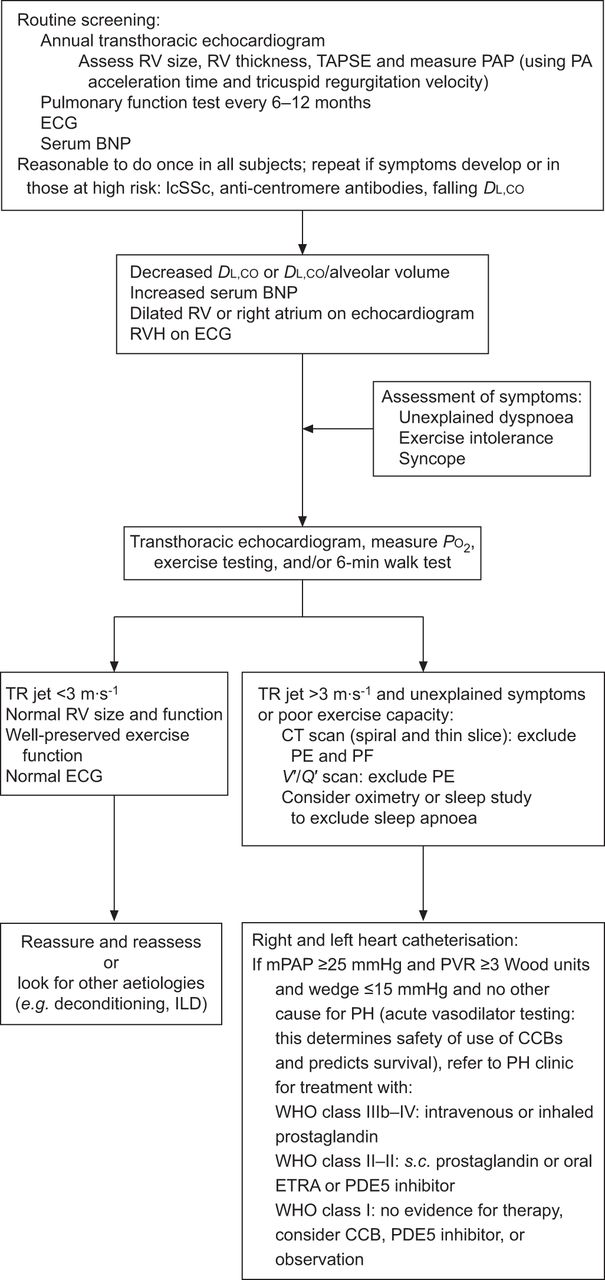
An algorithm for the early diagnosis of pulmonary arterial hypertension in systemic sclerosis. RV: right ventricle; TAPSE: tricuspid annular plane systolic excursion; PAP: pulmonary artery pressure; PA: pulmonary artery; BNP: B-type natriuretic protein; lcSSc: limited cutaneous systemic sclerosis; DL,CO: diffusing capacity of the lung for carbon monoxide; RVH: right ventricular hypertrophy; PO2: oxygen tension; TR: tricuspid regurgitation; CT: computed tomography; PE: pulmonary embolism; PF: pulmonary fibrosis; V′/Q′: ventilation/perfusion ratio; ILD: interstitial lung disease; mPAP: mean PAP; PVR: pulmonary vascular resistance; CCB: calcium channel blockers; WHO: World Health Organization; ETRA: endothelin receptor antagonist; PDE: phosphodiesterase. Reproduced from [110] with permission from the publisher.
Lung transplantation
Lung transplantation can be lifesaving in patients with SSc end-stage lung disease. Although there is a perception amongst some physicians that patients with scleroderma will have poor post-transplant outcomes due to concomitant gastro-oesophageal disease, renal disease or skin fibrosis, 2- and 5-yr outcomes are similar to patients transplanted for other conditions (72% and 55%, respectively) [153, 154]. Relative contraindications include significant skin breakdown from severe cutaneous disease, a creatinine clearance <50 mL·min−1, severe reflux disease and aspiration and cardiac involvement with arrhythmias [153]. With increasing awareness of airway complications and poor outcomes in patients with gastro-oesophageal reflux (GOR) and oesophageal dysmotility problems, aperistalsis, as determined by oesophageal manometry, is an absolute contraindication for lung transplantation in most lung transplant programmes; an unfortunate situation for patients with SSc as several patients with advanced stages of SSc-ILD are confronted with aperistalsis. In patients with SSc-PAH, transplantation remains an option in those who fail therapy. Reported outcomes are similar to those seen in IPF and IPAH [154].
Airway disease
Airway disease is rare in SSc when compared to other CVDs (e.g. rheumatoid arthritis). Older studies investigating airways disease in smoking and nonsmoking patients with SSc found that functional evidence of airflow obstruction could be attributed to a history of smoking [155], and nonsmoking SSc patients had airways disease at rates similar to healthy controls [156]. In one study, cylindrical bronchiectasis was observed in 59% of patients with SSc who were screened with HRCT; the significance of these results is unclear, especially with the high degree of reflux and aspiration in these patients [157].
Pleural involvement
Pleural involvement in SSc is rare, with effusions reported in 7% of patients; more often in those with dcSSc [158]. The prevalence increases to 15% when scleroderma with overlap syndromes are included [159]. Spontaneous pneumothorax is a rare complication [160], and typically occurs in those with ILD.
INDIRECT PULMONARY COMPLICATIONS
Oesophageal disease and GOR
Oesophageal disease and GOR are common in SSc, and are reported in >50–90% of patients [157, 161, 162] and are risk factors for lung injury [163]. The subgroup of SSc-ILD patients have a higher incidence of oesophageal involvement with more severe motor impairment, lower pressures in the lower oesophageal sphincter and a higher frequency of GOR episodes that reach the proximal oesophagus [164, 165]. There is a correlation between the degree of DL,CO impairment and the degree of GOR and oesophageal motor impairment [161, 164]. Followed over time, patients with severe oesophageal motor disturbances had a faster deterioration in their DL,CO and a higher frequency of ILD on HRCT [164]. In spite of these suggestive data, not all studies have found a correlation between GOR and ILD [166].
Infection
Pulmonary infections are relatively common in patients with SSc and can be responsible for significant morbidity and excess mortality [167]. Patients with SSc are at increased risk to manifest respiratory infection because of host susceptibility factors, including: the factors associated with the underlying autoimmune disease, aspiration risks because of oesophageal dysfunction, treatment with immune modulating agents, and respiratory muscle weakness. Thus, when patients with SSc manifest new pulmonary symptoms, both routine and opportunistic lung infections should be considered for appropriate diagnostic and therapeutic interventions [168].
Drug toxicity
Most of the medications used in the treatment of SSc have been associated with the development of pulmonary toxicity, including methotrexate (MTX), CYC, azathioprine and MMF. However, the diagnosis of drug-induced lung disease is challenging, given the nonspecific nature of the presenting signs and symptoms, chest imaging pattern and biopsy findings. The temporal relationship of the new/superimposed pulmonary manifestation to the initiation of the medications may help in differentiating the drug toxicity from the direct pulmonary manifestations of SSc. A more comprehensive list of medications with their reported pulmonary toxicities can be found online (www.pneumotox.com).
MTX is used in a number of autoimmune disorders including scleroderma. When pulmonary toxicity develops it is characterised by the development of dyspnoea, cough and fever over a period of a few weeks (though more acute and chronic presentations do occur) [169]. On chest imaging, superimposed/new interstitial infiltrates/GGOs in the lung fields may be present. While MTX usually causes hypersensitivity pneumonitis (granulomatous pneumonitis), a cellular (lymphoplasmacytic) infiltrate with or without granulomas, and acute and organising diffuse alveolar damage can also be seen [170].
CYC is one of the immune modulating agents used for SSc-ILD and recent clinical trials support its use [64, 66]. Both acute and chronic pulmonary toxicity have been described with CYC. Acute toxicity typically occurs after 1–6 months of exposure and is potentially reversible with cessation of therapy and corticosteroids. Chronic toxicity is reported to occur after months to years with the development of lung fibrosis and pleural thickening. This form of toxicity is typically irreversible and may be progressive despite the cessation of the drug [171].
Azathioprine has been rarely associated with pulmonary toxicity ranging from diffuse alveolar damage to lung fibrosis, and these effects have been reported to be dose-related [172]. MMF is rarely associated with pulmonary toxicity [55].
Malignancy
There are conflicting epidemiological data regarding an increased risk of malignancy in SSc patients. In studies that have reported an elevated risk, lung cancer (including bronchoalveolar cell carcinoma and adenocarcinoma) was the highest reported malignancy, representing nearly a third of all cancers [173]. The development of lung cancer appears to occur more frequently in the setting of ILD [174].
Respiratory muscle weakness
Skeletal muscle involvement is seen in SSc and can lead to global weakness [175, 176]. Respiratory muscle dysfunction with subsequent hypercapnea has been reported [177, 178].
Restrictive lung disease from skin and subcutaneous chest wall involvement
Restrictive lung disease from severe thoracic cutaneous involvement has been reported [179].
SUMMARY
ILD and PH are the leading causes of death in SSc patients. Due to other manifestations of their disease, as well as the asymptomatic nature of lung involvement in its early stages, pulmonary involvement often goes undiagnosed. Clinicians need to have a low threshold to evaluate for ILD and PH in these patients. Once diagnosed, therapy or enrolment in a treatment trial is recommended. While lung transplant is an option in selected patients with advanced lung disease, other non-pharmacological treatment interventions and goal-oriented measures that are not addressed in this review may influence outcome for patients with pulmonary manifestations of SSc.
Footnotes
Provenance
Submitted article, peer reviewed.
Statement of Interest
A. Fischer is an investigator for Scleroderma Lung Study II, and has received speaker and consultant fees from Actelion and is on medical advisory boards for Actelion. T. Bull has received two investigatorinitiated grants from United Therapeutics and has served as a consultant for Actelion and Lung Rx.
- Received September 13, 2012.
- Accepted December 5, 2012.
- ©ERS 2013
REFERENCES









