





To the Editor:
We report the case of an elderly male referred with pulmonary opacities and extensive mediastinal lymphadenopathy suspicious for lung cancer. He was found to have Rosai–Dorfman disease in a lymph node and concomitant immunoglobulin (Ig)G4-related disease in the lung and kidney.
An 83-yr-old male was referred with a pulmonary consolidation in the left lung with extensive lymphadenopathy without a pathological diagnosis. His past medical record showed gout for which he used colchicine only occasionally. Initially, he was referred to an internist with complaints of loss of appetite, weight loss and fatigue. His sense of taste was diminished and he had a dry mouth. No dyspnoea was present. He had a longstanding, non-productive cough. Until recently, he was remarkably fit for his age. He had quit smoking 45 yrs earlier after 30 pack-yrs.
Physical examination was unremarkable except for a firm lymph node in the left supraclavicular region and minor pulmonary crackles basally on both sides. Spirometry results were normal. Initial laboratory analysis showed a normal complete blood count, and slightly elevated creatinine (168 μmol·L−1) and C-reactive protein (35 mg·L−1).
A chest radiograph showed a hazy consolidation radiating from the left hilum to the periphery of the lung suggesting lung cancer. On computed tomography, diffuse opacification in the left lower lobe was seen with concomitant hilar, mediastinal (paratracheal and subcranial) and left supraclavicular lymphadenopathy.
Fibreoptic bronchoscopy in our institution again revealed no endobronchial lesions. Pathological examination of lavage fluid from the left lower lobe showed no malignant cells, only the presence of lymphocytes. Culture revealed no microorganisms (including mycobacteria).
Subsequently, an 18F-fluorodeoxyglucose (FDG) positron emission tomography was performed. The lesion in the left lung and the lymph nodes all showed strongly increased FDG uptake, again suggesting a metastatic lung cancer. In addition, the abdominal aortic wall also showed slight FDG uptake (fig. 1).

a) Positron emission tomography scan showing 18F-fluorodeoxyglucose uptake in the right supraclavicular, hilar and mediastinal lymph nodes and in the left lung. b) A computed tomography scan showing consolidation in the left lower lobe with enlarged mediastinal lymph nodes.
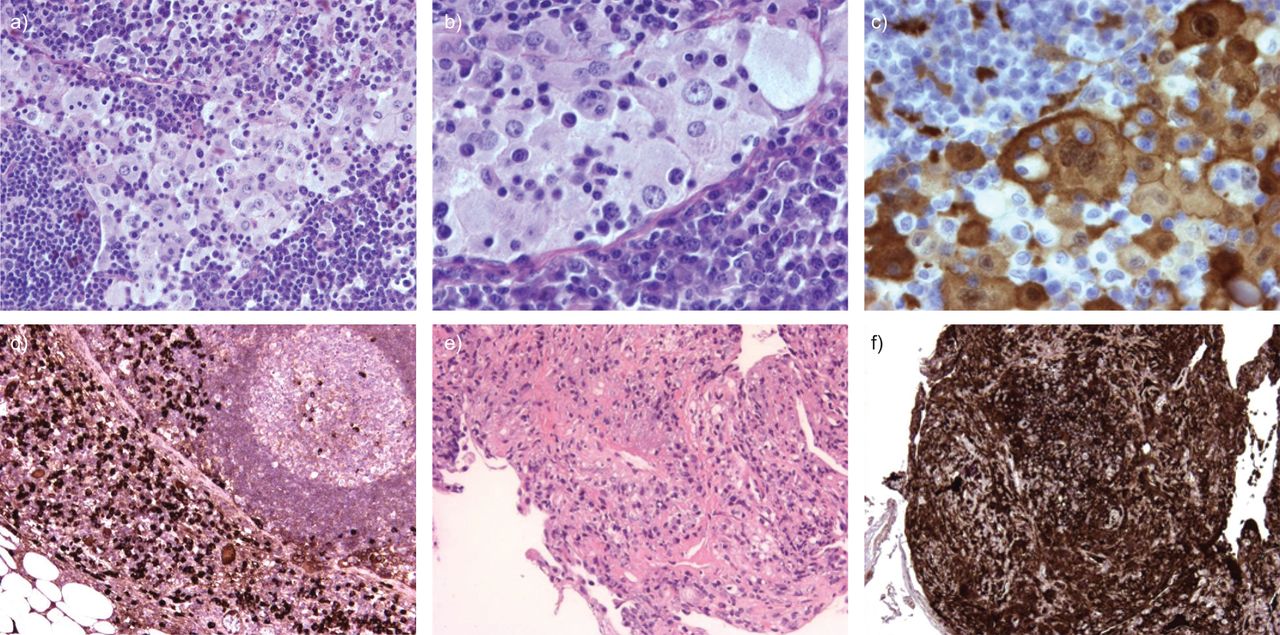
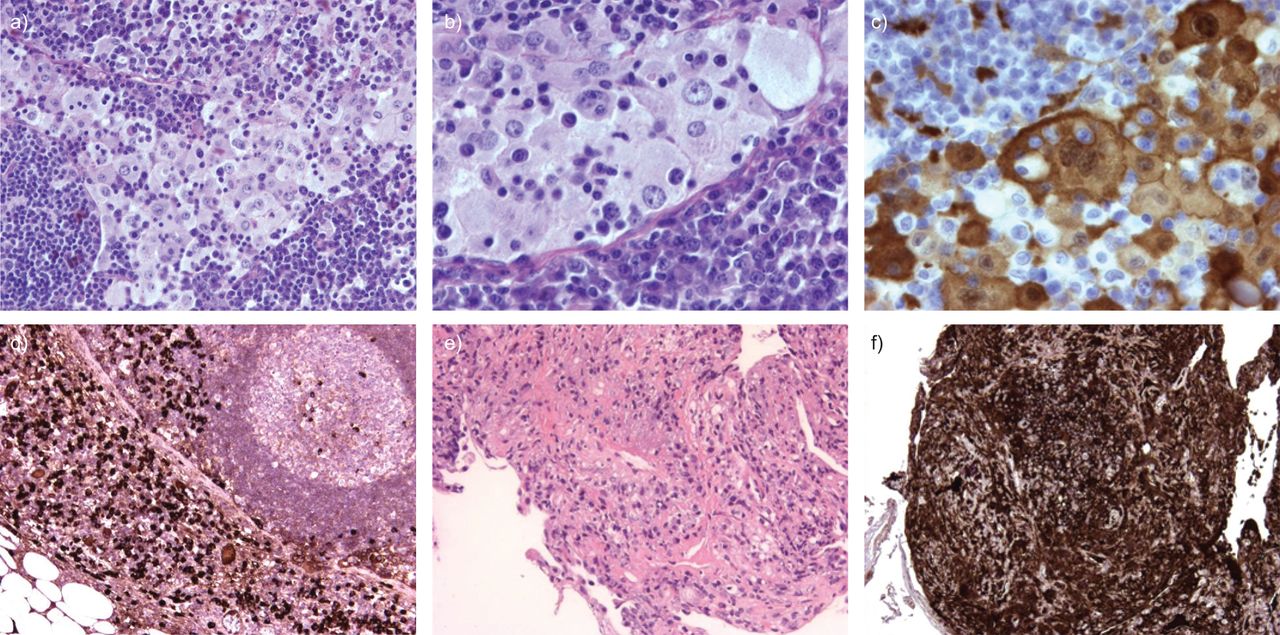
It was decided to remove the left supraclavicular lymph node for diagnostic and staging purposes. Pathological examination revealed extensive sinus infiltration with histiocytes, extensive cytoplasm with emperipolesis and prominent nuclei. These histiocytes were S-100 positive on immunohistochemistry. Between the histiocytes were many lymphocytes and even more plasma cells. Further immunohistochemistry showed that the plasma cells were IgG positive, with >20% being IgG4 positive (fig. 2a–d). This predominant histiocyte pattern is very compatible with Rosai–Dorman disease, although the high number of IgG4 positive plasma cells is not classically consistent with Rosai–Dorfman disease.
{kind=link}
{kind=link}
a–d) Pathological examination of left supraclavicular lymph node. e and f) Transbronchial lung biopsies of the left lower lobe. a) Haematoxylin and eosin staining showing extensive dilated sinuses with accumulation of histiocytes, lymphocytes and plasma cells. b) Further magnification with emperipolesis of lymphocytes by histiocytes. c) S-100 staining showing positive histiocytes. d) Immunoglobulin (Ig)G4 staining showing several IgG4 positive plasma cells. e) Haematoxylin and eosin staining showing inflammation with some fibrosis and infiltration of the lung parenchyma with plasma cells. The majority of these plasma cells are IgG4 positive. f) IgG4 staining.
Subsequent laboratory tests showed an elevated total serum IgG of 54 g·L−1, with an elevated IgG4 of 52.5 g·L−1 (upper limits of normal for total IgG and IgG4 are 16 g·L−1 and 1.4 g·L−1, respectively).
A second bronchoscopy was performed with transbronchial lung biopsies of the left lower lobe. Pathological examination showed inflammation with some fibrosis, few histiocytes and infiltration of the lung parenchyma with plasma cells, the majority were IgG4 positive (fig. 2e and f). This pattern is consistent with pulmonary lesions with IgG4-related disease. Although some histiocytes were observed, the morphological pattern was different from the pattern observed in the lymph node with Rosai–Dorfman disease.
Because of the renal dysfunction a kidney biopsy was performed that showed interstitial nephritis with abundant IgG4 plasma cells. Bone marrow examination revealed normal haematopoiesis, some polyclonal IgG4 positive plasma cells and no signs of Rosai-–Dorfman disease.
It was concluded that two rare disease patterns co-existed in our patient, Rosai–Dorfman disease in the lymph node and IgG4-related disease in the lung parenchyma, with proven renal involvement and probable aortic wall and salivary gland (Sjögren-like) involvement. Treatment with prednisone 60 mg once daily was started. Within several weeks his clinical condition improved, with clearance of all pulmonary lesions and improvement of the renal function tests.
Rosai–Dorfman disease, also known as sinus histiocytosis with massive lymphadenopathy, is a rare disease characterised by (mostly cervical) lymph node enlargement and fever, with a distinct cellular pattern of the lymph nodes [1]. The histopathological findings are massive sinus infiltration with histiocytes that show a large amount of cytoplasm in which lymphocytes are engulfed (emperipolesis; Rosai–Dorfman cells) that show immunohistochemical staining for S-100 protein without CD1a expression. Some hypothesise that Rosai-Dorfman disease is a reactive pattern in response to an (viral) infection, but the true cause remains unknown [2]. Extranodal disease involvement (pulmonary, kidney and skin) has been described previously [3].
Rosai–Dorfman disease is most frequent in young adults. The clinical course is unpredictable, and may be fluctuating and often prolonged. In the majority of cases, however, the disease is self-limiting. Most patients do not require treatment with steroids [1].
IgG4-related disease is a relatively newly discovered entity [4, 5]. It is characterised by an inflammatory and fibrosing infiltrate rich in IgG4-positive plasma cells and often elevated IgG4 serum concentrations. The most well-known affected organ is the pancreas, a disease formerly known as auto-immune pancreatitis. However, all internal organs may be affected, for instance the salivary glands (Sjögren-like syndrome), retroperitoneum (retroperitoneal fibrosis), kidney (interstitial nephritis), lymph nodes and the aorta (inflammatory aneurysm) [4]. The aetiology and pathogenesis are not known. Early IgG4-related disease is very steroid responsive with quick and longstanding remissions, and decreases in serum IgG4 levels. In cases with extensive fibrotic lesions, remission after therapy is less likely.
IgG4-related disease is also known for its pulmonary involvement. Nodular, bronchovascular, pleural, alveolar interstitial and round-shaped ground-glass opacity presentations have been described previously [6–8]. All presentations showed an infiltration of IgG4 positive plasma cells with sclerosing inflammation in the respective tissues, with concomitant increased plasma levels of IgG4. Pulmonary involvement frequently coincides with extrapulmonary disease, such as pancreas, retroperitoneum and kidney. In most cases, steroid therapy is effective.
Hypergammaglobulinaemia has long been considered part of Rosai–Dorfman disease. Rosai–Dorfman disease was only recently associated with increased IgG4 cell populations [9, 10]. This suggests a possible overlap or even a common cause between Rosai–Dorfman disease and IgG4-related disease. One could even speculate that Rosai–Dorfman disease might be a reactive pattern to IgG4-related disease. The underlying pathophysiology of this overlap and the clinical significance with respect to treatment and prognosis is not yet clear.
In summary, we present a patient who was admitted for suspected lung cancer but who was eventually diagnosed with two rare disease patterns: Rosai–Dorfman disease and IgG4-related disease. IgG4-related disease is a relatively underdiagnosed condition in which increasing scientific interest exists. There is evidence of an overlap between these entities (Rosai–Dorfman disease might be a reactive pattern to IgG4-related disease), but the cause and implications of this overlap are not known.
Footnotes
Provenance
Submitted article, peer reviewed.
Statement of Interest
None declared.
- ©ERS 2012









