





Abstract
Pulmonary arterial hypertension (PAH) and chronic thromboembolic pulmonary hypertension (CTEPH) are two of the key subgroups of pulmonary hypertension. They are characterised by different risk factors. PAH can be associated with mutations in the gene encoding bone morphogenetic protein receptor type II (BMPR2), HIV infection, congenital heart disease, connective tissue disease (such as systemic sclerosis), and exposure to particular drugs and toxins including fenfluramine derivatives. In contrast, CTEPH can be associated with anti-phospholipid antibodies, splenectomy and the presence of a ventriculo-atrial shunt or an infected pacemaker.
The first-line therapies used to treat PAH and CTEPH also differ. While medical therapy tends to be used for patients with PAH, pulmonary endarterectomy is the treatment of choice for patients with CTEPH.
However, there are possible common mechanisms behind the two diseases, including endothelial cell dysfunction and distal pulmonary artery remodelling. Further research into these similarities is needed to assist the development of targeted pharmacological therapies for patients with inoperable CTEPH and patients who have persistent pulmonary hypertension after endarterectomy.
- Chronic thromboembolic pulmonary hypertension
- epidemiology
- pathophysiology
- pulmonary arterial hypertension
The classification of pulmonary hypertension (PH) was recently updated at the 4th World Symposium on Pulmonary Hypertension [1]. This new classification delineates six key subgroups of PH: pulmonary arterial hypertension (PAH); pulmonary veno-occlusive disease and/or pulmonary capillary haemangiomatosis; PH due to left heart disease; PH due to lung diseases and/or hypoxia; chronic thromboembolic pulmonary hypertension (CTEPH); and PH with unclear or multifactorial mechanisms. This review will focus on the pathophysiology of PAH and CTEPH.
PAH
Mechanisms of disease
When obstruction of small pulmonary arteries, which is characteristic of PAH, was first described in the 1950s [2], it was thought that vasoconstriction and thrombosis were the key mechanisms of disease. However, it is now known that vasoconstriction is the dominant feature in <10% of patients at PAH diagnosis; these rare patients are characterised by long-term vasodilator response to calcium channel blockers [3]. Furthermore, thrombosis alone cannot explain PAH, although it may contribute to its pathogenesis. Remodelling of small pulmonary arteries (<500 μm diameter) via the proliferation of smooth muscle and endothelial cells is now recognised to play a major role in the pathogenesis of PAH [4]. This abnormal proliferation includes hypertrophy of the media and intima, and the formation of tumour-like lesions from endothelial cells in regions of pulmonary artery bifurcation (plexiform lesions). Such proliferation is likely to be an important target for future pharmacological therapies.
Risk factors
Familial PAH accounts for ∼4% of PAH cases [5]. Three-quarters of these patients harbour a mutation in the gene encoding bone morphogenetic protein receptor type II (BMPR2). Patients with such mutations are described as having heritable PAH, which tends to develop at a younger age than idiopathic PAH and often presents with a more severe clinical and haemodynamic phenotype [6]. Germline mutations in BMPR2 have also been found in 11–40% of apparently sporadic and idiopathic cases of PAH [7]. Furthermore, 10–20% of individuals affected by appetite suppressant-induced PAH carry a BMPR2 mutation [8, 9]. The receptor encoded by BMPR2 belongs to the transforming growth factor (TGF)-β superfamily, which plays a key role in vascular cell proliferation [10, 11]. However, the BMPR2 mutation has a penetrance of only ∼10–20%. This suggests that this genetic predisposition increases susceptibility to PAH, but that additional risk factors need to be present to induce pulmonary vascular dysfunction at the level of endothelial cells and smooth muscle cells [12]. The role of additional risk factors is highlighted by the recently updated clinical classification of PAH [1], which emphasises that PAH can be induced by various factors, including drugs or toxins.
Dysfunctional pathways and corresponding treatment options
Several therapies have been developed for PAH, which target three key pathways (fig. 1⇓): 1) the endothelin-1 pathway, targeted by endothelin receptor antagonists such as bosentan, sitaxsentan and ambrisentan; 2) the nitric oxide pathway, acted upon by phosphodiesterase type 5 inhibitors such as sildenafil; and 3) the prostacyclin pathway, targeted by prostacyclin analogues such as epoprostenol, treprostinil and iloprost [3, 14]. These therapies can improve symptoms and exercise capacity. Although they have some anti-proliferative properties, they do not reverse abnormal cell proliferation in vivo and, as such, do not represent a cure for PAH. Thus, growth factors such as platelet-derived growth factor, epidermal growth factor and fibroblast growth factor, which are involved in abnormal proliferation, form targets for future novel therapies. However, no convincing data have yet been published to support the use of drugs targeting these novel pathways.
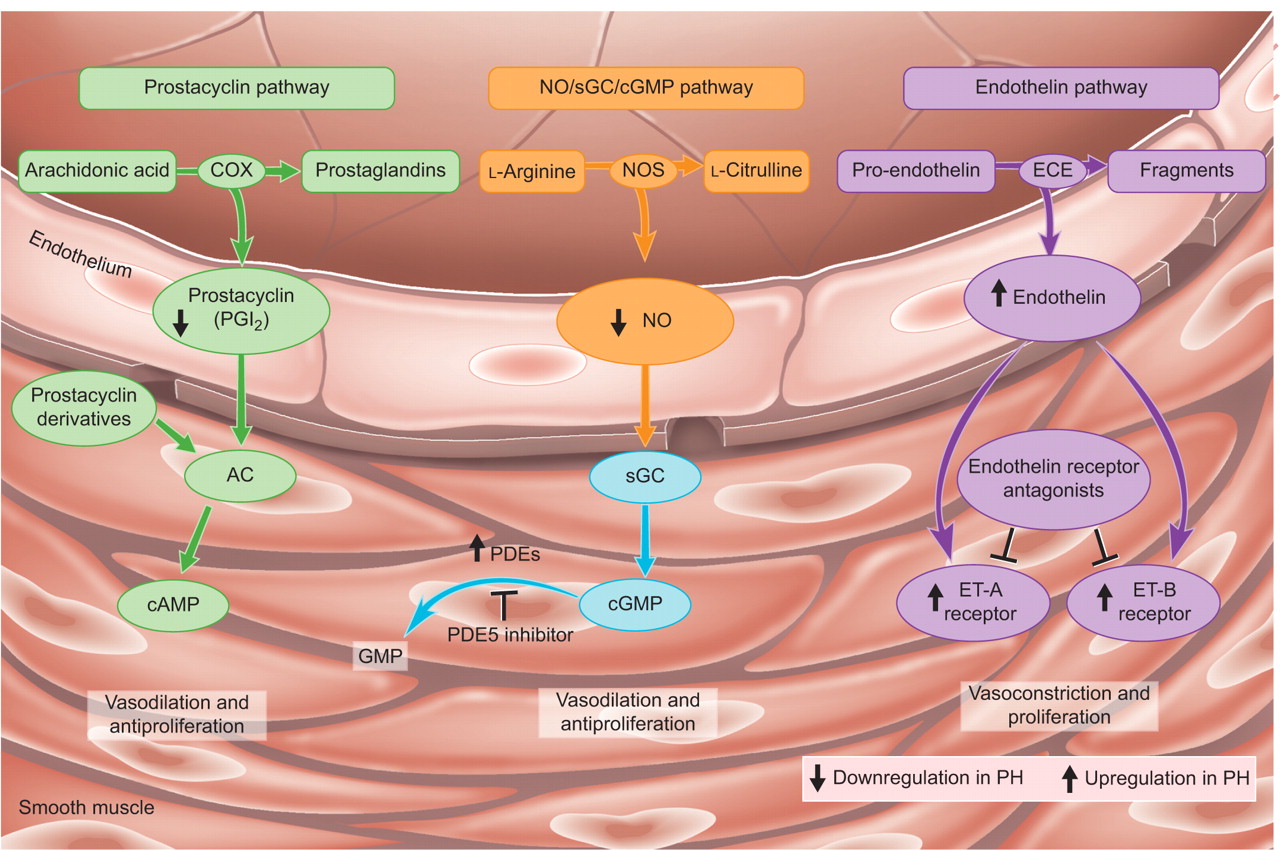
The key pathways and classes of drugs that have been approved for the treatment of pulmonary arterial hypertension. These may also have benefits in patients with chronic thromboembolic pulmonary hypertension, although further research is necessary to establish this. NO: nitric oxide; sGC: soluble guanylate cyclise; cGMP: cyclic guanosine monophosphate; COX: cyclo-oxygenase; NOS: NO synthase; ECE: endothelin converting enzyme; AC: adenylate cyclase; cAMP: cyclic adenosine monophosphate; ET: endothelin; GMP: guanosine monophosphate; PDE: phosphodiesterase; PH: pulmonary hypertension. Reproduced from [13] with permission from the publisher.
CTEPH
Mechanisms of disease
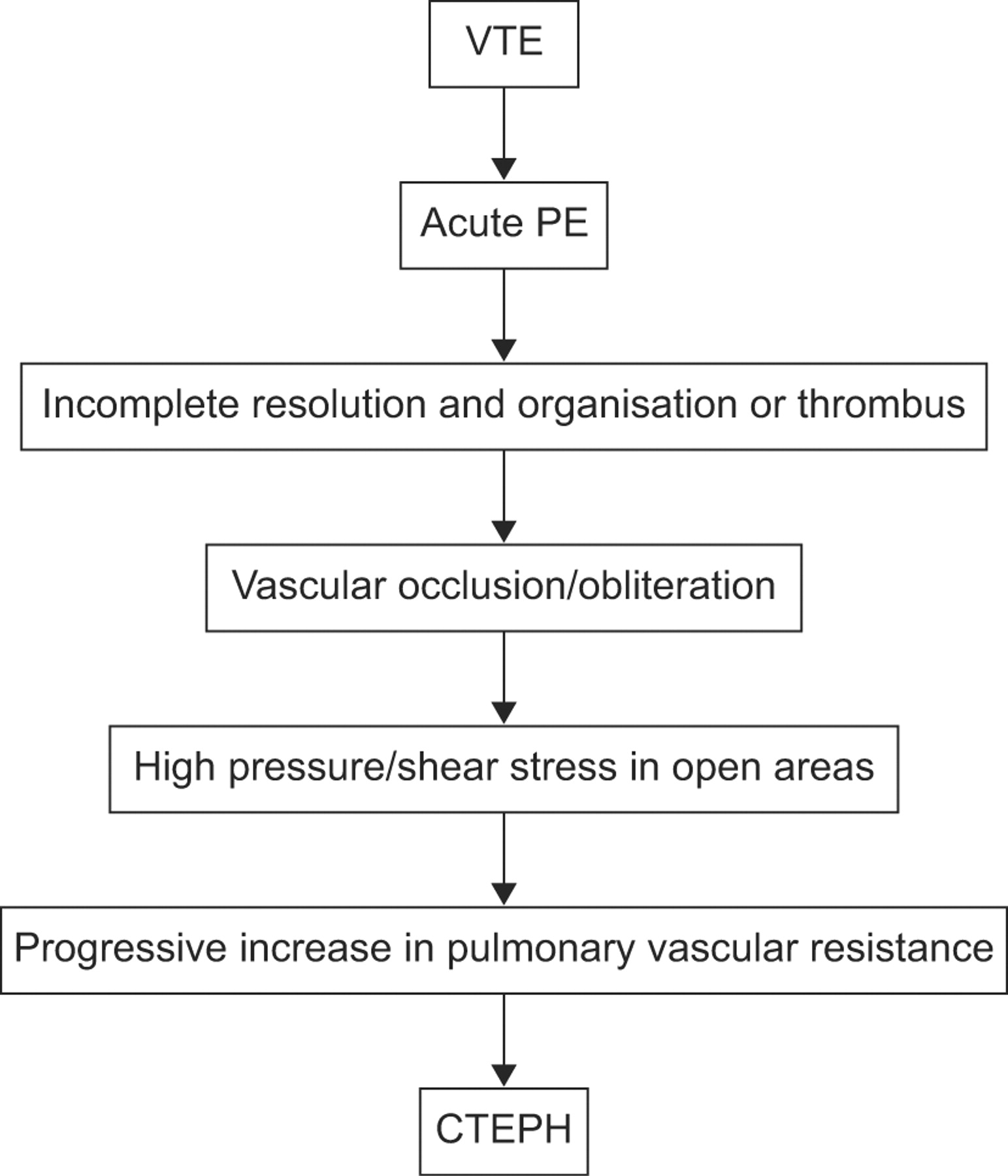
Unlike PAH where vascular remodelling tends to occur in small pulmonary arteries, CTEPH is mainly associated with prominent obstructions in larger vessels. The pathophysiology of CTEPH remains unclear. The commonly accepted explanation (the embolic hypothesis) is that CTEPH is the result of single or recurrent pulmonary embolism (PE) arising from sites of venous thrombosis (fig. 2⇓) [15]. However, it has been suggested that CTEPH may also be caused by in situ thrombosis in the lung as a result of primary arteriopathy and endothelial dysfunction similar to that seen in PAH [15–17]. This may help to explain why up to 63% of patients with CTEPH have no history of acute PE [18]. Interestingly, progressive remodelling may occur in small pulmonary arteries in occluded and nonoccluded territories, which supports the possible relevance of pulmonary arteriopathy in CTEPH [17].

{kind=link}
{kind=link}
The embolic hypothesis of chronic thromboembolic pulmonary hypertension (CTEPH). VTE: venous thromboembolism; PE: pulmonary embolism.
Incidence and prevalence
Historically, the incidence of CTEPH was estimated at 0.1–0.5% in patients surviving acute PE [19]. However, a more recent study found the cumulative incidence of CTEPH to be 3.1% (95% CI 0.7–5.5) 1 yr after PE and 3.8% (95% CI 1.1–6.5) 2 yrs after PE [20]. The true incidence of CTEPH after acute PE is now thought to be between 0.5% and 2% [11].
Natural history
The natural history of CTEPH is difficult to characterise because there can be a period (the so-called “honeymoon” period) of months to years between the initiating event, which may be silent, and the onset of CTEPH symptoms [21]. Furthermore, patients may initially present with nonspecific symptoms, such as mild breathlessness and a general feeling of being out of shape. The consequence of this is that patients are often only diagnosed at the late stage of the disease, when they present with the typical symptoms of progressive dyspnoea on exertion and general, clinical deterioration paralleling the loss of right ventricular functional capacity.
Risk factors
Independent predictors of CTEPH include the presence of a ventriculo-atrial shunt or infected pacemaker (OR 76, 95% CI 8–10,351) and splenectomy (OR 18, 95% CI 2–2,438) [22]. Chronic inflammatory disorders, such as osteomyelitis and inflammatory bowel disease are also associated with an increased risk of CTEPH (OR 67, 95% CI 8–8,832) [23]. Previous PE (OR 19.0, 95% CI 4.5–79.8), younger age (OR 1.8 per 10 yrs, 95% CI 1.2–2.6), a larger perfusion defect (OR 2.2 per decile decrement in perfusion, 95% CI 1.5–3.3), and idiopathic PE at presentation (OR 5.7, 95% CI 1.4–23.0) are associated with an increased risk of CTEPH after acute PE [20].
Anti-phospholipid antibodies are the most prevalent biological abnormality found in patients with CTEPH. Indeed, anti-phospholipid antibodies are more common in patients with CTEPH than in patients with PAH [24]. In addition, recent reports indicate that significantly higher levels of plasma factor VIII have been found in patients with CTEPH than in healthy controls (41% versus 5%; p<0.0001) [25]. This study also found plasma factor VIII levels to be significantly higher in patients with CTEPH than in patients with PAH (41% versus 22%; p<0.022). There is, however, no evidence of a link between the risk of CTEPH and antithrombin deficiency, protein C deficiency, protein S deficiency or factor V Leiden [24].
An abnormal fibrinolytic response may also help to explain why some patients develop CTEPH after an acute PE. One study of genetic mutations underlying fibrinogen structural variants reported a prevalence of dysfibrinogenaemia of 15% (95% CI 3–27) in 22 patients with CTEPH [26]. However, it should be noted that the prevalence of dysfibrinogenaemia in the general population is unknown.
Treatment options
On multidetector helical computed tomography of the chest or on pulmonary angiography, acute PE appears as a floating clot in the pulmonary artery surrounded by contrast. Therefore, the clots are very simple to remove if necessary. CTEPH, however, appears as abnormal thickening of the pulmonary arterial wall and is characterised by the presence of fibrous scars that occlude the pulmonary artery lumen. It should also be noted that the bronchial arteries are often markedly enlarged in CTEPH due to bronchial arterial angiogenesis. Pulmonary endarterectomy is associated with excellent results [27] and is the treatment of choice for patients with CTEPH [28]. However, there are risks associated with pulmonary endarterectomy, and CTEPH is inoperable in at least 20–40% of patients [29] because of distal disease or comorbidities [27].
Therefore, there is a need for effective pharmacotherapies for the treatment of CTEPH [30]. PAH therapy is sometimes considered as a bridge to surgery in patients with severe but accessible CTEPH [31]. PAH drugs belonging to the three traditional classes have been studied in CTEPH [32–34], and novel drugs that tackle vasoconstriction and proliferation [30, 35, 36], as well as therapies based on growth factors, may also have benefits in patients with CTEPH. However, no drug is currently approved for the medical management of CTEPH and further studies are needed to establish the efficacy of these agents in that setting [32].
CONCLUSIONS
PAH and CTEPH are characterised by different risk factors. PAH can be associated with mutations in the BMPR2 gene, HIV infection, congenital heart disease, connective tissue disease (such as systemic sclerosis) and exposure to particular drugs and toxins including fenfluramine derivatives. In contrast, CTEPH can be associated with anti-phospholipid antibodies, splenectomy and the presence of a ventriculo-atrial shunt or an infected pacemaker. The first-line therapies that are used to treat PAH and CTEPH are also different. While medical therapy tends to be used in patients with PAH, pulmonary endarterectomy is the treatment of choice in patients with CTEPH. However, there are possible common mechanisms behind the two diseases, including endothelial cell dysfunction and distal pulmonary artery remodelling. Further research into these similarities will aid the development of targeted therapies for patients with inoperable CTEPH and patients who have persistent pulmonary hypertension after endarterectomy.
Statement of interest
M. Humbert has relationships with drug companies including Actelion, Bayer Schering, GlaxoSmithKline, Novartis, Pfizer and United Therapeutics. In addition to being investigator in trials involving these companies, relationships include consultancy service and membership of scientific advisory boards.
Provenance
Publication of this peer-reviewed article was supported by Bayer Schering Pharma AG, Germany (principal sponsor, European Respiratory Review issue 115).
Acknowledgments
Medical writing support was provided by C. Hill (Oxford PharmaGenesis Ltd, Oxford, UK) on behalf of Bayer Schering Pharma AG (Berlin, Germany). This article is based on a presentation given at a symposium supported by Bayer Schering Pharma AG at the 2009 European Society of Cardiology meeting held in Barcelona, Spain.
- Received November 23, 2009.
- Accepted December 4, 2009.
- © ERSJ Ltd
References









