





Abstract
Endothelial cells regulate vascular tone largely by the actions of endothelin-1. Endothelin-1 is a potent vasoconstrictor, with effects that are dependent on the receptors to which it binds as well as their location. Endothelin-1 dysregulation is implicated in pathological conditions, including those of the pulmonary vasculature and the kidney. In this review, we describe the physiology and actions of endothelin-1 in lung and renal tissues and discuss therapies that disrupt these interactions in disease states. We provide an overview of the current clinical progress of these targeted agents and provide perspectives on the treatment of pulmonary and renal diseases with endothelin receptor antagonists.
Endothelial cells (ECs) play a key role in the regulation of vascular tone by the production of mediators with vasoactive activity, such as prostacyclins, nitric oxide (NO) and endothelins (ETs). These mediators have opposing roles on the whole vascular bed, including smooth muscle cells, matrix and fibroblasts, as well as platelets and inflammatory pathways. ET-1 is produced by ECs and is involved in severe disorders, including those of the pulmonary vasculature and the kidney. In this review, we describe the current knowledge in ET science, clinical progress and the prospects for the treatment of pulmonary and renal diseases.
ET-1 AND ENDOTHELIN RECEPTORS
The ETs comprise a family of three 21-amino acid peptides: ET-1, ET-2 and ET-3 [1]. ET-1 is the principal isoform in the human cardiovascular system and remains one of the most powerful constrictors of human vessels that has been discovered [2, 3]. ET-1 is also a potent mitogen that can induce proliferation in multiple cell types, including vascular smooth muscle cells [4].
ET-1 is synthesised within ECs from a biologically inactive precursor, big ET-1, by hydrolysis predominantly mediated by the endothelin-converting enzyme (ECE-1) localised to the plasma membrane [5]. A second enzyme, ECE-2, optimally functions at an acidic pH [6], which may occur under pathophysiological conditions [7]. Approximately 25% of big ET-1 is released to circulate within the plasma and may undergo conversion to ET-1 by ECE that is present on smooth muscle; in this manner, big ET-1 could function as a long-range signalling molecule. Finally, chymase, which is present in mast cells, can mediate an alternative conversion pathway transforming big ET-1 to mature peptide. This is interesting because mast cells are increased in cardiovascular disease, including in atherosclerotic lesions [7].
ET-1 is continuously released from ECs by a constitutive pathway (fig. 1⇓) [8], causing vasoconstriction [9]. This release is balanced by the release of vasodilators to maintain vascular tone. Release of ET-1 can also occur from a second regulated pathway in which Weibel–Palade bodies (EC-specific storage granules) degranulate in response to external mechanical or chemical stimuli [10].

Schematic diagram of endothelin (ET)-1 release in the human cardiovascular system. ET-1 is synthesised by endothelin-converting enzymes (ECE-1 or ECE-2) within vesicles and continuously released via the constitutive pathway to interact with smooth muscle ET receptors, mainly the ETA subtype, and contribute to the maintenance of vascular tone. In some, but not all, human vessels, a small population of ETB receptors can also mediate constriction. ET-1 is also synthesised by ECE-1 and stored in Weibel–Palade bodies until released following an external physiological or pathophysiological stimulus (regulated pathway) to produce further vasoconstriction. Activation of ETB receptors by ET-1 limits the constrictor response by the release of vasodilators such as nitric oxide (NO). ETB receptors, mainly in the kidney and lungs, remove ET-1 from the circulation and also have a beneficial role in limiting any rise in ET-1 resulting from ETA receptor blockade. ET-1 is also released from endocardial endothelial cells lining the chambers of the heart to produce positive inotropic action effects on myocytes mediated by ETA receptors.
ETs activate two distinct G protein-coupled receptors, ETA and ETB [11–14]. Binding to the different receptors produces opposite effects on the vascular bed. ETA receptors are abundantly expressed on vascular smooth muscle cells throughout the human cardiovascular system, including the lungs and kidney, and are believed to mediate vasoconstriction, proliferation, apoptosis and fibrosis. A low density of ETB receptors (<15%) is also present on smooth muscle cells but contributes little to vasoconstriction in either normal or diseased vessels [7]. ETB receptors are predominantly expressed on ECs throughout the human vasculature, including the lungs [13, 15]. Upon activation by ET-1, ETB receptors mediate the beneficial release of endothelium-derived relaxing factors, such as NO, in a feedback mechanism that limits ETA vasoconstriction. High densities of ETB receptors are present in the lung, kidney and liver, where this subtype functions as a clearing receptor to internalise the ligand-receptor complex and remove ET-1 from circulation [16–19]. As a result, the plasma half-life of ET-1 is comparatively short [20].
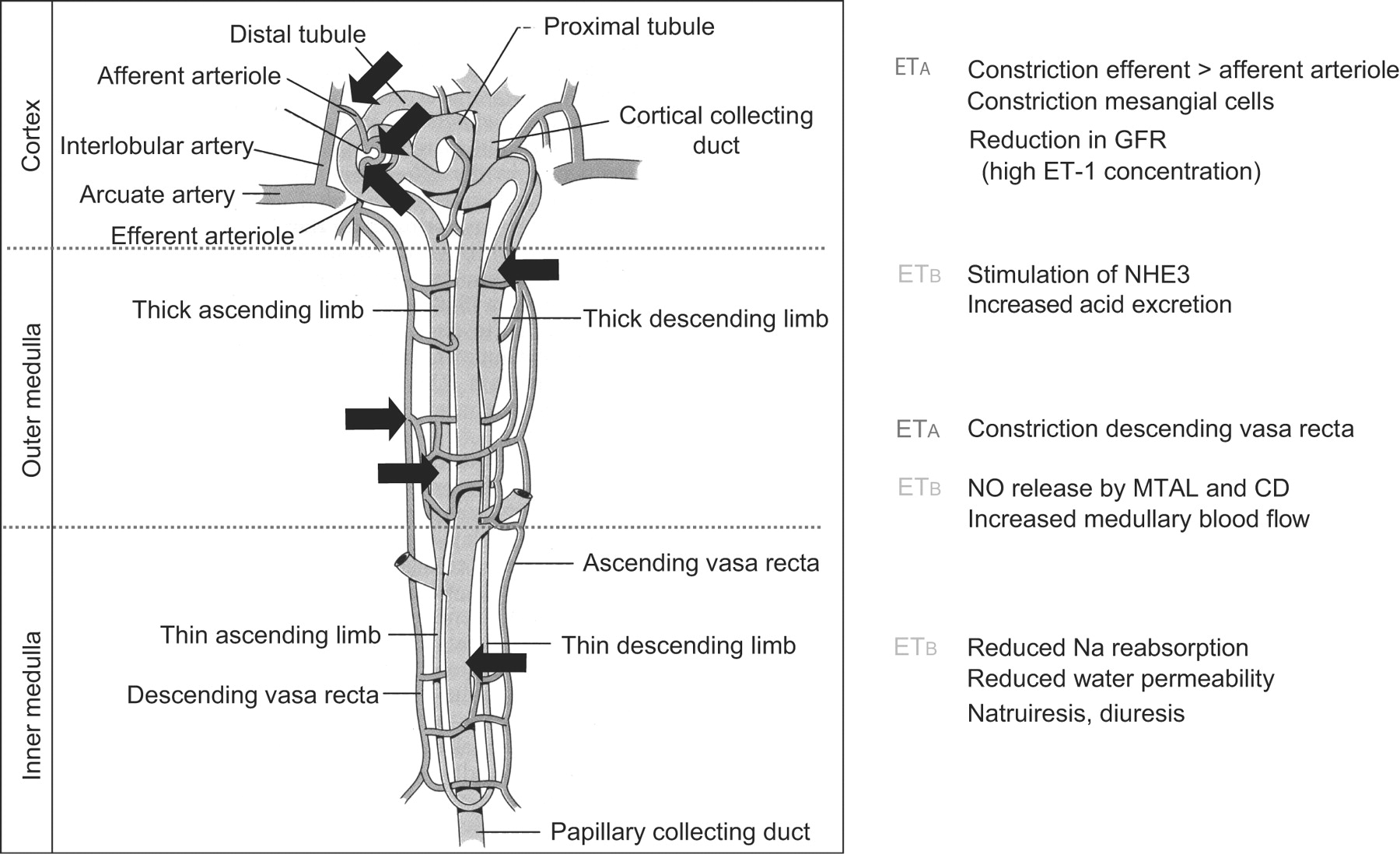
In marked contrast to vascular smooth muscle, ETB receptors predominate in the human kidney, comprising ∼70% of the receptors present in both the cortex and medulla [21, 22]. ETB receptors are present on ECs throughout the renal vasculature, consistent with a role in mediating dilatation by endothelium-derived vasodilators as well as clearing receptors, thus, removing ET-1 from circulation (fig. 2⇓). ETB receptors are also present on epithelial cells throughout the tubular epithelium, particularly the inner medullary collecting duct where the major action of ET-1 is to promote diuresis and natriuresis (table 1⇓).

The effects of endothelin (ET)-1 in a healthy kidney. ETA and ETB: G protein-coupled receptors activated by ET; GFR: glomerular filtration rate; NHE3: sodium-hydrogen exchanger 3; NO: nitric oxide; MTAL: medullary thick ascending limb; CD: collecting duct.
Response to endothelin(ET)-1 in the kidney by receptor subtype
The majority of the ETA receptors are localised to renal vessels of the medulla and cortex, including the main renal and arcuate arteries and adjacent veins at the cortico-medullary junction, as well as intra-renal resistance vessels and arterioles supplying the glomerulus [22]. ETA is also present on other cell types including mesangial cells and podocytes [21].
In addition to removing ET-1 from circulation, the human kidney is also a source of substantial amounts of ET-1, with ET-1 staining localised to the cytoplasm of ECs lining intra-renal vessel arteries, veins and resistance vessels, as well as ECs within the glomerulus [23]. Intra-renal ET-1 primarily acts in an autocrine or paracrine manner with its main physiological actions being ETA-mediated constriction in the vasculature and ETB-mediated diuresis and natriuresis in the renal medulla. In renal disease, ET-1 causes vasoconstriction as well as extracellular matrix production, mesangial-cell proliferation and inflammation, mainly via ETA receptors [21]. Interestingly, the renal vasculature is almost an order of magnitude more sensitive to ET-1 than any other vascular bed [24, 25].
RATIONALE FOR ET-1 RECEPTOR BLOCKADE: INCREASE OF ET-1 EXPRESSION AND RECEPTOR SUBTYPES
High levels of ET-1 are present in multiple disease states, making the ET pathway an attractive target for pharmacological intervention. Increased ET-1 activity is present in patients with pulmonary arterial hypertension (PAH). In PAH, the blood flow through the pulmonary arterial circulation is reduced, leading to increased pulmonary vascular resistance and ultimately to right heart failure [26]. The condition is ultimately fatal; the 1-yr mortality rate remains high at ∼15% with modern therapy [27]. A key review of the pathology, classification and treatment of PAH has recently been published [28].
Plasma levels of ET-1 are increased in PAH and correlate with the severity of disease, as well as its prognosis [29, 30]. Explanted lungs of patients with PAH show increased ET-1 production [31], as do lung biopsy specimens of paediatric patients with PAH [32]. Patients with PAH have reduced clearance of ET-1 in the pulmonary vasculature [33]. Furthermore, binding sites for both types of ET-1 receptors are upregulated in the distal pulmonary arteries of patients with PAH [34].
The ET system has also been implicated in kidney disease [5]. Plasma ET-1 levels increase as renal function declines in chronic kidney disease (CKD) [35, 36] and may contribute to hypertension. Upregulated renal ET-1 production is also observed in CKD [37], reflected in higher urinary ET-1 excretion [36, 38]. Animal data suggest that in CKD, the renal vasculature may be more sensitive to the vasoconstrictor effects of ET-1 than normal kidneys [39].
SELECTIVE (ETA) OR NONSELECTIVE (ETA/ETB) RECEPTOR BLOCKADE?
The various functions of each ET-receptor subtype ensure maintenance of vascular tone and function. Because ETA receptors in the pulmonary system are associated with multiple deleterious effects in PAH [40], inhibition is crucial. Nonselective ET-1 receptor antagonists (classified as blocking ETA and ETB receptors with similar potency) have reversed some of the observed pathological conditions associated with diseases, which may include inhibiting some functions of the ETB receptors on vascular smooth muscle, whose role in disease is poorly understood. However, because in healthy tissue the endothelial ETB receptor opposes and clears ET-1, dual blockade may not be desirable. Furthermore, the elevated levels of ET-1 that may result from ETB receptor blockade may not be desirable in PAH, where ET-1 levels are already dysregulated.
In the kidney, there are several reasons why ETA-selective antagonists might be superior to mixed blockade [21]. The pathogenic actions of ET-1 in the kidney (including vasoconstriction, extracellular matrix production and inflammation) are largely mediated by ETA receptors. Inhibition of the renal medullary ET-1/ETB system causes sodium retention and hypertension because of its role in systemic fluid and electrolyte homeostasis. Additionally, ETB receptors counteract the vasoconstrictive effects of ETA receptors and are essential for removing ET-1 from the systemic circulation.
Therefore, nonselective antagonism would be expected to block the natriuretic, diuretic and vasodilatory effects of the ETB receptors in the kidney and the antihypertensive effects of ETB receptors in the circulatory system, in addition to opposing the detrimental effects of ETA receptors [21]. Additionally, reduced ETB-mediated clearance would probably increase plasma ET-1 levels [21]. In agreement, pre-clinical data show that selective ETA antagonism may be preferential to nonselective blockade in CKD [41, 42].
Although the role of the ET system has been widely studied, little is known about the potential interactions between the renal and the pulmonary circulation. However, PAH patients have significant renal dysfunction, which is likely to adversely affect prognosis [43]. Additionally, renal crisis in the scleroderma population is, at least in part, mediated by the ET system. Thus, there is the potential for cross-talk between the pulmonary and kidney vasculature via the ETA and ETB receptors [43].
CLINICAL EVIDENCE FOR ET-1 BLOCKADE IN PAH
ET-receptor antagonists have been studied most extensively in PAH; several compounds have been approved for the treatment of the disease. ET-receptor agonists have also been investigated for the treatment of cardiovascular disorders, systemic hypertension, and chronic and acute heart failure [40].
Several agents have been developed for clinical use and are classified as either nonselective (including bosentan and tezosentan), modestly ETA selective (including ambrisentan and darusentan) or highly ETA selective (sitaxentan). The distinction between antagonists that are ETA selective and those that block both receptors is not precise, but theoretically the former should display at least 100-fold selectivity for the ETA subtype so that concentrations can be achieved to block >90% of the ETA but <10% ETB (fig. 3⇓) [44]. In practice, it is difficult to achieve a steady-state concentration with precision, even in experimental clinical studies; selectivities >1,000-fold may be necessary and are currently only achieved with sitaxentan. Blocking the ETB subtype increases the level of circulating plasma ET-1 [17], which can serve as a biomarker of ETB receptor antagonism. In contrast with the experience in pulmonary hypertension, all clinical trials of heart failure have used mixed antagonists or doses of modestly selective ETA antagonists high enough to block the ETB receptor (evidenced by a rise in plasma ET-1), with disappointing results [45].

The reported degree of selectivity of endothelin (ET) receptor antagonists for ETA receptors, shown by manufacturer classification. ▓: ETA; □: ETA and ETB. #: nonselective.
Nonselective antagonists
As the first orally available, dual-receptor antagonist in its class, the clinical experience with bosentan is the most extensive (table 2⇓). The first randomised, double-blind, placebo-controlled study of bosentan examined 32 patients with functional class III or IV idiopathic PAH or PAH associated with the scleroderma spectrum of diseases [46]. Exercise capacity, as assessed by the 6-min walk distance (6MWD), was significantly improved (by a mean of 70 m) compared with baseline after 12 weeks of bosentan treatment; the improvement with bosentan was significantly greater than that observed with placebo treatment. Haemodynamically, bosentan significantly improved cardiac index and reduced mean pulmonary arterial pressure (P̄pa) and pulmonary vascular resistance (PVR). New York Heart Association/World Health Organization (NYHA/WHO) functional class was significantly improved in patients receiving bosentan treatment. An asymptomatic increase in hepatic aminotransferase was observed in two patients treated with bosentan; the number and nature of adverse events (AEs) did not differ between placebo and bosentan groups.
Randomised controlled trials with endothelin receptor antagonists in pulmonary arterial hypertension
A larger, double-blind, placebo-controlled trial examined 213 patients with WHO functional class III or IV idiopathic PAH or PAH associated with connective tissue disease (PAH-CTD) who received 16 weeks of bosentan treatment [49] at two doses of 125 and 250 mg b.i.d. The improvement in 6MWD observed with bosentan treatment was significantly greater than the deterioration observed with placebo treatment. Time to clinical worsening was defined as time to death, lung transplantation, hospitalisation for PAH, lack of clinical improvement or worsening leading to discontinuation, need for epoprostenol therapy or atrial septostomy. In this study, the risk of clinical worsening was significantly reduced with bosentan compared with placebo treatment [49]. Abnormal hepatic function, syncope and flushing were more common with bosentan treatment. Elevated transaminase levels were more frequently observed at the highest dose of bosentan (250 mg b.i.d.), leading to drug discontinuation in 2% versus 0% in the active and placebo arm, respectively.
More recently, the effects of bosentan were examined in a 26-week study of 168 patients with earlier, functional class II PAH of multiple causes (including PAH associated with anorexigen use and congenital heart disease (CHD)) [51]. Co-primary end-points were change in 6MWD and PVR. PVR was significantly improved with bosentan treatment but 6MWD was not, perhaps owing to the higher baseline 6MWD (435 m) of these less impaired patients, which may have made improvement difficult to detect. This study strongly supports the need for early intervention because vascular remodelling might be more feasible before disease progression [51].
Long-term bosentan treatment appears beneficial for survival, although to determine this equivocally would require ethically unacceptable extended studies with a placebo comparator arm. Kaplan–Meier survival estimates were 96% at 1 yr and 89% at 2 yrs for 169 patients with WHO functional class III or IV idiopathic or heritable PAH or PAH-CTD who received first-line bosentan therapy and continued in open-label extension studies. At the end of 1 and 2 yrs, 85% and 70% of patients, respectively, were alive and on bosentan monotherapy; additional or alternative therapy for patients was permitted at the discretion of the treating physician [54]. Survival in patients with functional class III idiopathic PAH treated with bosentan (n = 139) was compared with a historical control group treated with epoprostenol (n = 346) [55]. Improvements in exercise capacity and haemodynamics observed at 4 months were maintained at 1 yr. Kaplan–Meier survival estimates after 1 and 2 yrs were 97% and 91%, respectively, for bosentan and 91% and 84%, respectively, for epoprostenol. Notably, the epoprostenol group had more severe disease, but survival estimates were similar in matched cohorts of 83 patients. Strikingly similar observations for exercise capacity and haemodynamics were noted in a retrospective analysis of 103 consecutive NYHA functional class III/IV patients with idiopathic PAH treated with bosentan at a single centre [56]. Survival at 1 and 2 yrs was 90% and 87%, respectively; however, 44% of patients required additional prostanoid treatment. Elevated liver enzymes were noted in all of the long-term bosentan trials.
Bosentan has effectively treated other forms of PAH as well; supporting the idea that ET is a pathogenically relevant mediator in PAH of various aetiologies. Although the pathogenesis and mechanism of development of PAH associated with HIV are uncertain, 16 patients with functional class III and IV PAH-HIV who enrolled in an uncontrolled, prospective, 16-week study showed improved exercise capacity, quality of life and haemodynamics with bosentan treatment. Control of HIV infection was unaffected by treatment [57]. Furthermore, in a longer term (29±15 months) analysis of 59 patients with functional class II through to IV PAH-HIV (including 12 patients from the original study) treated with first-line bosentan, exercise capacity remained stable while haemodynamics were further improved; in 10 patients, haemodynamics stabilised and improved to WHO functional class I [58]. 11 consecutive patients with NYHA functional class III and IV portopulmonary hypertension (PPHT) and Child A cirrhosis with stable liver function who were treated for >1 yr with bosentan improved 6MWD/exercise capacity with a significant increase in peak oxygen uptake [59]. PVR fell significantly while other haemodynamic measures showed improvement (including cardiac output, P̄pa, right atrial pressure, mixed venous oxygen saturation and mean systemic artery pressure), although a significant worsening of arterial oxygenation was observed [60]. A retrospective study of 31 patients with PPHT who received either iloprost (n = 13) or bosentan (n = 18) revealed statistically significant differences in survival rates with bosentan treatment (94%, 89% and 89% at 1, 2 and 3 yrs, respectively) compared with iloprost treatment (77%, 62% and 46% at 1, 2 and 3 yrs, respectively) [61].
ETA-selective antagonists
Clinical success has also been demonstrated in PAH using selective ETA receptor antagonists. Sitaxentan (research code TBC-11251) is up to 6,500 times more selective for the ETA than ETB receptors [62]. Two randomised, double-blind, placebo-controlled studies examined the efficacy and safety of short-term treatment (STRIDE-1 and -2). In STRIDE-1, 178 NYHA functional class II through to IV patients with idiopathic PAH, PAH-CTD or PAH-CHD received placebo, 100 mg sitaxentan q.d. or 300 mg sitaxentan q.d. for 12 weeks [52]. Both doses of sitaxentan significantly improved 6MWD, although the primary end-point, the change in peak volume of oxygen consumed, was not significantly improved with 100 mg sitaxentan. Functional class and haemodynamic measures (cardiac index and PVR) were significantly improved with sitaxentan treatment. Liver function abnormalities were reported for 300 mg of sitaxentan but not for 100 mg of sitaxentan. Other frequent AEs included headache, peripheral oedema, nausea, nasal congestion and dizziness. Increased international normalised ratio or prothrombin time was the most frequently reported laboratory AE. In the second trial, 247 patients with WHO functional class II through to IV idiopathic PAH, PAH-CTD or PAH-CHD received placebo, 50 mg sitaxentan q.d. or 100 mg sitaxentan q.d. for 18 weeks; an open-label bosentan arm was included [53]. The primary end-point of 6MWD was significantly improved in the 100 mg sitaxentan but not the 50 mg sitaxentan group and was also significantly improved in the bosentan group. Significant improvement in WHO functional class only occurred in the 100 mg sitaxentan group. Elevated hepatic transaminases occurred for all treatment groups, including placebo, at a comparable rate.
In the STRIDE-2 extension trial for sitaxentan, PAH patients who completed the double-blind trial continued to receive either sitaxentan (n = 145) or bosentan (n = 84) for 1 yr. At 1 yr, sitaxentan therapy showed significant benefit over bosentan with respect to discontinuation of monotherapy, clinical worsening, elevated hepatic transaminases and survival (96% versus 88%, respectively) [63, 64], which, according to the authors, was related to the ETA receptor selectivity of sitaxentan [64]. However, this study does not provide a true head-to-head comparison because patients randomised in the bosentan arm were receiving open-label bosentan. In addition, placebo patients were randomised after the double-blind phase of the trial to receive sitaxentan 100 mg q.d. or bosentan 125 mg b.i.d., creating a potential bias in the analysis. Long-term treatment with 100 mg sitaxentan is associated with a 76% survival rate at 3 yrs, with 60% of the patients remaining on monotherapy [65].
Interestingly, sitaxentan has been investigated as an alternative ET-receptor antagonist for patients discontinuing bosentan therapy [66]. In a randomised, double-blind study of 48 patients with idiopathic PAH, PAH-CTD or PAH-CHD who had discontinued bosentan for inadequate efficacy (n = 35) or safety concerns (n = 13), patients received 50 or 100 mg sitaxentan q.d. In patients who discontinued for efficacy reasons, 100 mg sitaxentan improved 6MWD in five (33%) out of 15 patients compared with two (10%) out of 20 patients treated with 50 mg sitaxentan. Similar results were seen for the Borg dyspnoea index and WHO functional class. Of 12 patients who discontinued bosentan because of hepatotoxicity, only one patient developed elevated liver enzymes (after 13 weeks of sitaxentan therapy).
Of 42 PAH-CTD patients from the STRIDE-1 study, data from those who received 100 or 300 mg sitaxentan (n = 33) were pooled and compared with data from placebo-treated patients (n = 9) in a post hoc subset analysis [67]. After 12 weeks of treatment, mean 6MWD was significantly improved with sitaxentan treatment compared with the deterioration observed in the placebo group; parallel improvements in quality of life and haemodynamics were also observed. In 41 patients who entered the blinded extension study and received either 100 or 300 mg sitaxentan q.d. for a median treatment duration of 26 weeks, the proportion of patients having functional class I or II disease was significantly greater at the end of the study compared with the start of active therapy. Elevated liver enzymes were recorded in two patients.
Ambrisentan is another selective ETA receptor agonist; reports of the affinity of binding to ETA versus ETB receptors are variable but range up to 4,000-fold [68, 69]. The results of two concurrent, double-blind, placebo-controlled studies that randomised 202 and 192 patients with PAH, respectively, to placebo or ambrisentan (5 or 10 mg in ARIES-1; 2.5 or 5 mg in ARIES-2) orally once daily for 12 weeks followed by a 48-week extension study were published together [48]. The primary end-point of 6MWD was significantly increased in all ambrisentan groups. Time to clinical worsening was improved in ARIES-2 but not in ARIES-1, the difference lying primarily in a higher rate of deterioration in the placebo group observed in ARIES-2. Statistical improvements in WHO functional class, quality-of-life scores (assessed by the Short Form (SF)-36) and Borg dyspnoea score were observed in both studies. Liver abnormalities were not observed with ambrisentan treatment. Peripheral oedema, headache and nasal congestion were commonly associated with ambrisentan treatment.
In a subset of 280 patients from the concurrent trials who completed 48 weeks of treatment with ambrisentan monotherapy, the improvement in 6MWD observed at 12 weeks appeared to be maintained at 48 weeks [48]. Results of long-term response to ambrisentan in 383 patients with PAH (including those randomised to placebo in the controlled trials who received ambrisentan in an extension trial) have been presented recently [70]. Kaplan–Meier survival was estimated to be 94% and 88% at 1 and 2 yrs, respectively, whereas time to clinical worsening was estimated to be 88% and 72%, respectively. Of these patients, the incidence of aminotransferase levels more than three times the upper limit of normal (ULN) was 3.4%, which compares favourably with the 2.3% rate of increase observed in the placebo group during the first 12-week period.
Because ambrisentan was thought to have a lower risk of liver abnormalities than alternative ET-1-receptor antagonists, perhaps owing to the fact that it is propanoic-acid based, it was studied in 36 patients who had previously discontinued bosentan (n = 31), sitaxentan (n = 2) or both (n = 3) owing to liver function test abnormalities [71]. In this 12-week trial, no patients had drug-related aminotransferase levels more than three times the ULN that resulted in drug discontinuation, the incidence of which was the primary end-point, although one patient had a transient aminotransferase level more than three times the ULN that resolved following a temporary dose reduction. In 34 patients who continued treatment in an extension study for a median of 102 weeks, no additional patients reported aminotransferase levels more than three times the ULN, despite the fact that >50% of patients doubled their ambrisentan dose. Significantly improved 6MWD and Borg dyspnoea index compared with baseline was observed at 8 and 12 weeks. Furthermore, WHO functional class improved in 43% of patients, and significant improvements were observed in six out of eight domains of the SF-36 health survey and the composite physical health score.
Antagonists combined with other therapies
Combination trials of bosentan with prostanoids were based on improved exercise capacity and apparent safety in uncontrolled case series [72, 73] but have shown mixed results (table 3⇓). Bosentan was combined with epoprostenol, a continuously infused prostaglandin, in a double-blind, placebo-controlled prospective study [74]. 33 patients with idiopathic or familial PAH or PAH-CTD who were receiving epoprostenol were randomised to 16-week bosentan or placebo treatment. The bosentan plus epoprostenol group showed nonsignificant improvements in all measured haemodynamic parameters, failing to meet the primary end-point of total pulmonary resistance. The bosentan plus epoprostenol group had four withdrawals, including two deaths due to cardiopulmonary failure, one clinical worsening event and one AE compared with one withdrawal in the placebo plus epoprostenol group for an AE.
Combination trials with endothelin receptor antagonists(ERAs) in pulmonary arterial hypertension
Inhaled iloprost, a prostacyclin analogue, was similarly added to stable bosentan therapy in a 12-week, randomised, double-blind trial of 67 patients, of whom 63 (93%) had NYHA functional class III idiopathic and associated PAH [75]. Placebo or inhaled iloprost (5 μg) was added to stable bosentan monotherapy. The bosentan plus iloprost patients improved their 6MWD (p = 0.051) compared with bosentan plus placebo patients. Additionally, significantly more patients in the bosentan plus iloprost group improved NYHA status by one class than in the bosentan plus placebo group. Bosentan plus iloprost similarly demonstrated superiority in time to clinical worsening and haemodynamics. However, the clinical benefit observed could be attributable to iloprost alone because there was not a treatment arm in which iloprost replaced bosentan treatment. Another 12-week randomised controlled trial examining the addition of inhaled iloprost to bosentan treatment in 40 patients with idiopathic PAH was terminated early because the primary end-point of the 6MWD was not met [60].
The phosphodiesterase type 5 inhibitor sildenafil has been added safely to bosentan treatment after clinical deterioration following initial response to monotherapy in uncontrolled trials [59, 76]; controlled trials are underway. In the pivotal trial of tadalafil for the treatment of PAH, 53% of patients were on bosentan background therapy [77]. In contrast to treatment-naїve patients, improvements in 6MWD and WHO functional class were not significantly different in patients on background bosentan treatment who received 40 mg tadalafil versus placebo. Haemodynamics were “uniformly better” in treatment-naїve patients as well, although a small sample size was noted. Importantly, the sequential nature of treatment may have affected response; concurrent initiation of the two drugs in naїve patients may prove efficacious in adequately designed future trials. A case series of three patients with idiopathic PAH who sustained symptomatic and haemodynamic improvement after receiving sitaxentan and tadalafil in combination following bosentan failure may indicate promise with an ETA-selective agent [78].
Other combination trials are ongoing or planned with different compounds (table 2⇑). Conflicting results from recent combination trials raise some significant concerns. Many of the patients in combination trials were diagnosed >3 yrs before randomisation and were already receiving therapy. Although their disease is worsening, these patients are clearly selected with a lower risk of event because outcomes for “prevalent” patients are better than those for new (“incident”) patients [79]. However, the reason for this discrepancy is unclear, perhaps because of differences in initial treatment at nonspecialised centres, age, comorbidities or the prevalence of associated CTD.
Furthermore, the ideal strategy to treat disease is currently unclear. Are more aggressive approaches, such as initial combination therapy or quick escalation of therapy in response to unmet clinical goals, necessary for favourable long-term outcomes? More studies are necessary, yet study design has been extensively debated. A recent expert consensus document discussed the best tools to assess the effects of current or new compounds in PAH [28]. It was recommended that future trials use robust end-points, such as time to clinical worsening, to assess the efficacy of new treatment strategies.
CLINICAL EVIDENCE FOR ET-1 BLOCKADE IN KIDNEY DISEASES
In a randomised, double-blind crossover study of seven adult males with CKD (mean arterial pressure 103 mmHg, mean plasma creatinine 3.5 mg·dL−1), the experimental cyclic hexapeptide ET-1-receptor antagonist TAK-044 was used to briefly block both ETA and ETB receptors [78]. Each patient received placebo and 100 mg and 750 mg TAK-044 as 15-min intravenous infusions, with treatments separated by ≥7 days. TAK-044 is only modestly selective for the ETA receptor; the low concentration selectively blocks ETA but not ETB, whereas the higher concentration antagonises both ET-receptor subtypes.
Systemic and renal haemodynamics, as well as plasma immunoreactive ET-1, its precursor big ET-1 and the C-terminal fragment were measured before and after infusions [80]. No change in plasma ET-1 level was observed at the lower concentration of TAK-044. At the higher concentration, plasma ET-1 was increased two-fold without a concomitant increase in big ET-1 or the C-terminal fragment, implying that the increased plasma ET-1 in CKD does not result from de novo synthesis but rather reduced clearance of the peptide by ETB receptors.
Both doses of TAK-044 altered renal dynamics by lowering the effective filtration fraction [80]. Systemic vasodilatation and lowered blood pressure were observed at both doses of TAK-044, supporting the idea that ET-1 contributes to the maintenance of vascular tone in patients with CKD. Although the study was small, ET receptor antagonists may be beneficial for treating hypertension associated with CKD if this antihypertensive action is sustainable.
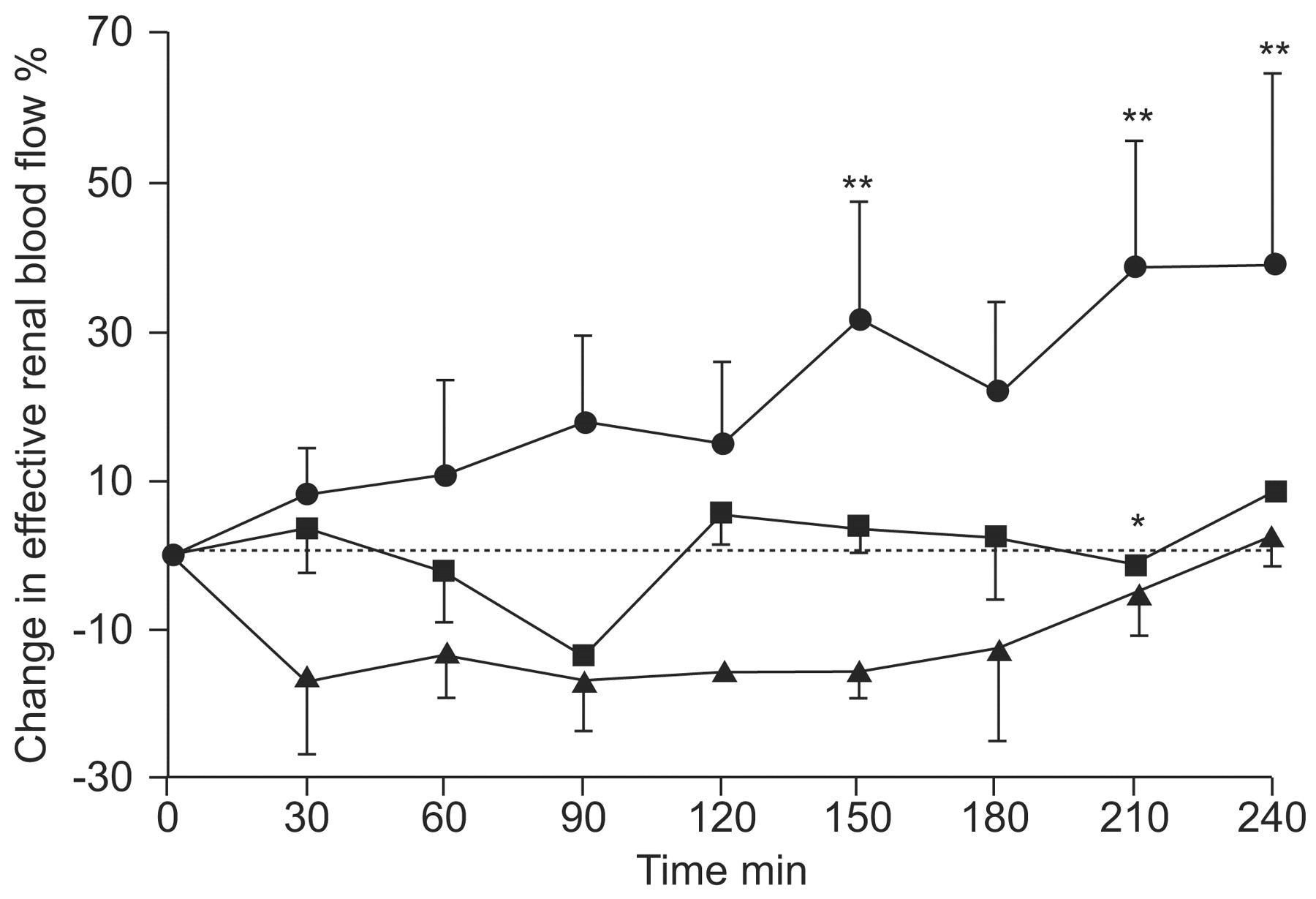
The first clinical study to assess ETA and ETB receptor antagonists in a head-to-head comparison used the experimental, but highly selective, peptide antagonists BQ-123 (ETA) and BQ-788 (ETB). In this randomised, double-blind, placebo-controlled, four-way crossover study, the antagonists were examined alone and in combination in eight hypertensive patients with chronic renal failure and eight matched healthy controls [42]. ETA receptor antagonism significantly reduced blood pressure in patients with chronic renal failure with or without ETB receptor antagonism, but the magnitude of change was significantly higher in the BQ-123-only group. BQ-123 also increased renal blood flow (fig. 4⇓) and renal vascular resistance and reduced proteinuria, consistent with a renoprotective action; these effects were not seen when BQ-123 was combined with BQ-788. Thus, ETB receptors maintain tonic renal vasodilatation. Consistent with this idea, ETB receptor blockade with BQ-788 produced both systemic and renal vasoconstriction in chronic renal failure patients and healthy controls. Surprisingly, no changes in sodium excretions were observed despite evidence from animal studies that ETB receptors mediate natriuresis [5]. Although these were acute studies, the results suggest an ETA selective antagonist would reduce blood pressure in patients with chronic renal failure and provide additional renoprotection. Additionally, this study suggests that therapeutic effects can be expected provided that the ETA receptors are blocked, regardless of ETB receptor blockade.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Change in effective renal blood flow. Selective endothelin (ET)A receptor antagonism increases renal blood flow in chronic kidney disease. •: ETA blockade; ▪: ETA + ETB blockade; ▴: ETB blockade. ETA >ETA + ETB. *: p<0.05 versus ETA antagonists; **: p<0.01 versus placebo. Reproduced from [42] with permission from the publisher.
In a randomised, placebo-controlled, double-blind, dose–response study in 11 healthy males, BQ-123 was infused intravenously in three separate doses (0.1, 0.2 and 0.3 mg·kg−1 for 1 h) [81]. Renal haemodynamics, tubular function, vasoactive hormones (plasma concentrations of renin, angiotensin II, atrial and brain natriuretic peptides and vasopressin) and blood pressure were measured in healthy males. Renal sodium excretion was increased despite a decrease in diastolic blood pressure and stimulation of the renin–angiotensin system. This suggests the natriuretic effect of BQ-123 overrides the sodium-retaining effect of increased activity in the renin–angiotensin system.
In a larger double-blind, randomised crossover study in which 22 patients with nondiabetic proteinuric CKD received, on two separate occasions, BQ-123 or placebo [82], ETA receptor antagonism significantly reduced blood pressure, proteinuria and arterial stiffness (a key determinant of cardiovascular risk) measured by pulse wave velocity. Importantly, all patients were treated with angiotensin-converting enzyme (ACE) inhibitors and/or angiotensin receptor antagonists (ARAs) titrated to the maximum tolerated dose throughout the study, so these effects occurred in addition to standard treatment. The reductions in proteinuria and arterial stiffness were partly independent of blood pressure because a subset of 10 patients who received 10 mg nifedipine as a third active control treatment demonstrated only comparable reductions in blood pressure and renal flow. Interestingly, BQ-123 produced a significant natriuresis in these patients, probably resulting from increased renal blood flow with ETA-receptor antagonism.
As in the previous trial, this was an acute study in a relatively homogenous group of patients. Patients with diabetes mellitus, vasculitis and renal vascular disease may benefit similarly from treatment. If the effects of ETA receptor antagonism can be maintained for longer periods, such treatment may confer cardiovascular and renal benefits in patients with CKD. Longer term trials are required to address these questions.
FUTURE PERSPECTIVES: A ROLE OF CARDIO-RENAL INTERACTIONS WITH SELECTIVE BLOCKADE?
Acute experimental clinical studies show that selective blockade of the ETA receptor preserves the natriuretic, antihypertensive and ET-1 clearance effects of ETB receptors, whereas nonselective blockage may reduce these benefits in patients with CKD. Furthermore, it may be beneficial to combine an ETA antagonist with ACE inhibitors or ARAs for CKD treatment. The sustained safety and efficacy of ET-receptor blockade observed in PAH support a rationale for long-term randomised trials with nonpeptide antagonists. Furthermore, this potential may include other renal diseases associated with elevated levels of ET-1 or renal vasoconstriction, such as acute renal failure, hepatorenal syndrome and cyclosporine therapy or characterised by cell proliferation, such as chronic renal failure [5].
Support statement
A. Davenport is supported by the British Heart Foundation. Editorial support was made possible by funding from Pfizer.
Statement of interest
J-L. Vachiéry has received fees for speaking and travel grants from Actelion Pharmaceuticals, GlaxoSmithKline, Pfizer, United Therapeutics and Bayer. He has received consultancy fees from GlaxoSmithKline, Pfizer, United Therapeutics and Elli Lilly as well as research grants from United Therapeutics. A. Davenport was the recipient of a Biotechnology and Biological Sciences Research Council Industrial Case PhD studentship. He has also received research grants from Pfizer, and reimbursement for attending symposia from Encysive and Pfizer.
Provenance
Publication of this article was made possible by funding from Pfizer, UK (article sponsor, European Respiratory Review issue 114).
Acknowledgments
The authors would like to thank T. Brake and J. Matsuura for providing editorial support (Complete Healthcare Communications, Chadds Ford, PA, USA).
- Received September 18, 2009.
- Accepted September 29, 2009.
- © ERSJ Ltd
References










Jump To
- Article
- Abstract
- ET-1 AND ENDOTHELIN RECEPTORS
- RATIONALE FOR ET-1 RECEPTOR BLOCKADE: INCREASE OF ET-1 EXPRESSION AND RECEPTOR SUBTYPES
- SELECTIVE (ETA) OR NONSELECTIVE (ETA/ETB) RECEPTOR BLOCKADE?
- CLINICAL EVIDENCE FOR ET-1 BLOCKADE IN PAH
- CLINICAL EVIDENCE FOR ET-1 BLOCKADE IN KIDNEY DISEASES
- FUTURE PERSPECTIVES: A ROLE OF CARDIO-RENAL INTERACTIONS WITH SELECTIVE BLOCKADE?
- Support statement
- Statement of interest
- Provenance
- Acknowledgments
- References
- Figures & Data
- Info & Metrics