





Abstract
Pulmonary arterial hypertension (PAH) commonly arises in patients with congenital heart disease (CHD). Greater numbers of patients with PAH associated with CHD (PAH-CHD) are now surviving into adulthood, many with increasingly complex cardiac defects. Patients with cardiac defects which result in left-to-right shunting are at risk of developing PAH, owing to the increased shear stress and circumferential stretch induced by increased pulmonary blood flow, which leads to endothelial dysfunction and progressive vascular remodelling and, thus, increased pulmonary vascular resistance. Although PAH-CHD shares similar lung histology with idiopathic PAH, differences do exist between these aetiologies. Management of PAH-CHD can involve surgical correction of the cardiac defect and/or treatment of the PAH, depending on the underlying cardiac defect and status of disease progression. Transplantation surgery can be curative but is not without limitations. The timing of intervention in patients with PAH-CHD is important, but the optimums are sometimes difficult to define, with limited robust data to inform management decisions. Uncontrolled studies suggest that prostacyclin analogues and phosphodiesterase type-5 inhibitors may have benefits in advanced pulmonary vascular disease. In the only randomised controlled trial dedicated to end-stage PAH-CHD, bosentan significantly reduced pulmonary vascular resistance and significantly increased 6-minute walk distance without compromising peripheral oxygen saturation, in patients with Eisenmenger syndrome. These data suggest that targeted therapies are beneficial in the PAH-CHD population, and warrant further research.
Pulmonary arterial hypertension (PAH) commonly arises in patients with congenital heart disease (CHD), and the prevalence of PAH associated with CHD (PAH-CHD) may be underestimated. Congenital heart defects occur in 0.8–1% of live births [1], and PAH is a major complication in children with left-to-right (systemic-to-pulmonary) shunts. Around 5% of adults with CHD develop PAH [2]. The prevalence of PAH-CHD has been estimated to be 1.6–12.5 cases per million adults [3], with 25–50% of these patients exhibiting the most serious form, Eisenmenger syndrome [3]. Among adults with PAH, 11.3% have PAH-CHD [4], but in children, around half of all cases of PAH have underlying CHD or Eisenmenger physiology [5]. Survival of children with untreated post-operative PAH-CHD is low [6, 7], and the presence of PAH is considered a significant predictor of problems in adult CHD patients undergoing surgery [8].
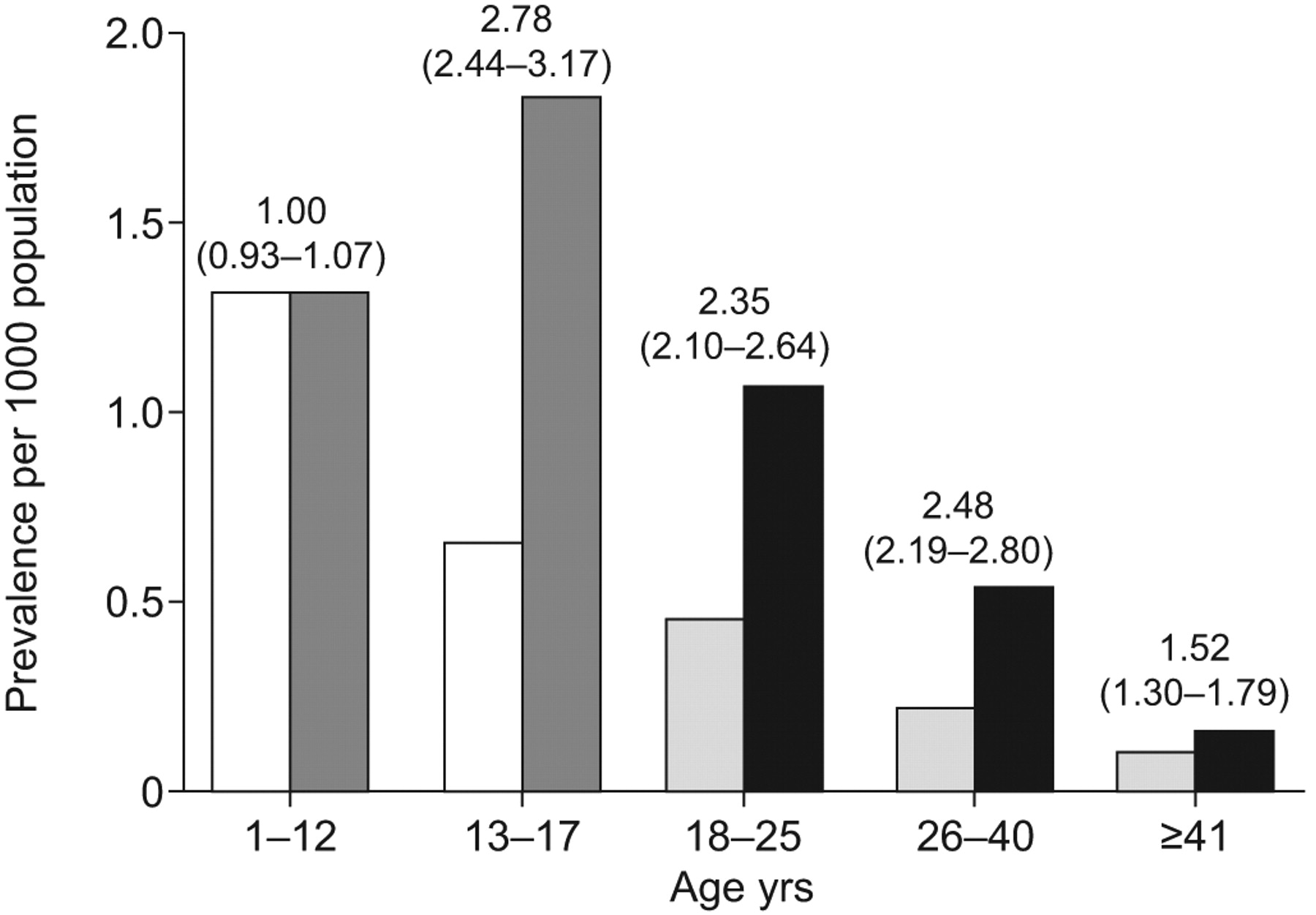
Although traditionally most cases of PAH-CHD have arisen in children, the demographics are changing. The prevalence of congenital heart defects in adults and adolescents was reported to increase between 1985 and 2000, probably due to more paediatric patients surviving into adulthood (fig. 1⇓) [1, 9]. Furthermore, with the progress in paediatric cardiology and cardiac surgery, there is likely to be a reduction in the number of adult patients with simple CHD (such as ventricular septal defects and patent ductus arteriosus), and an increase in those with complex CHD (such as single-ventricle physiology) who may develop pulmonary vascular lesions after undergoing palliative procedures in early childhood [10].
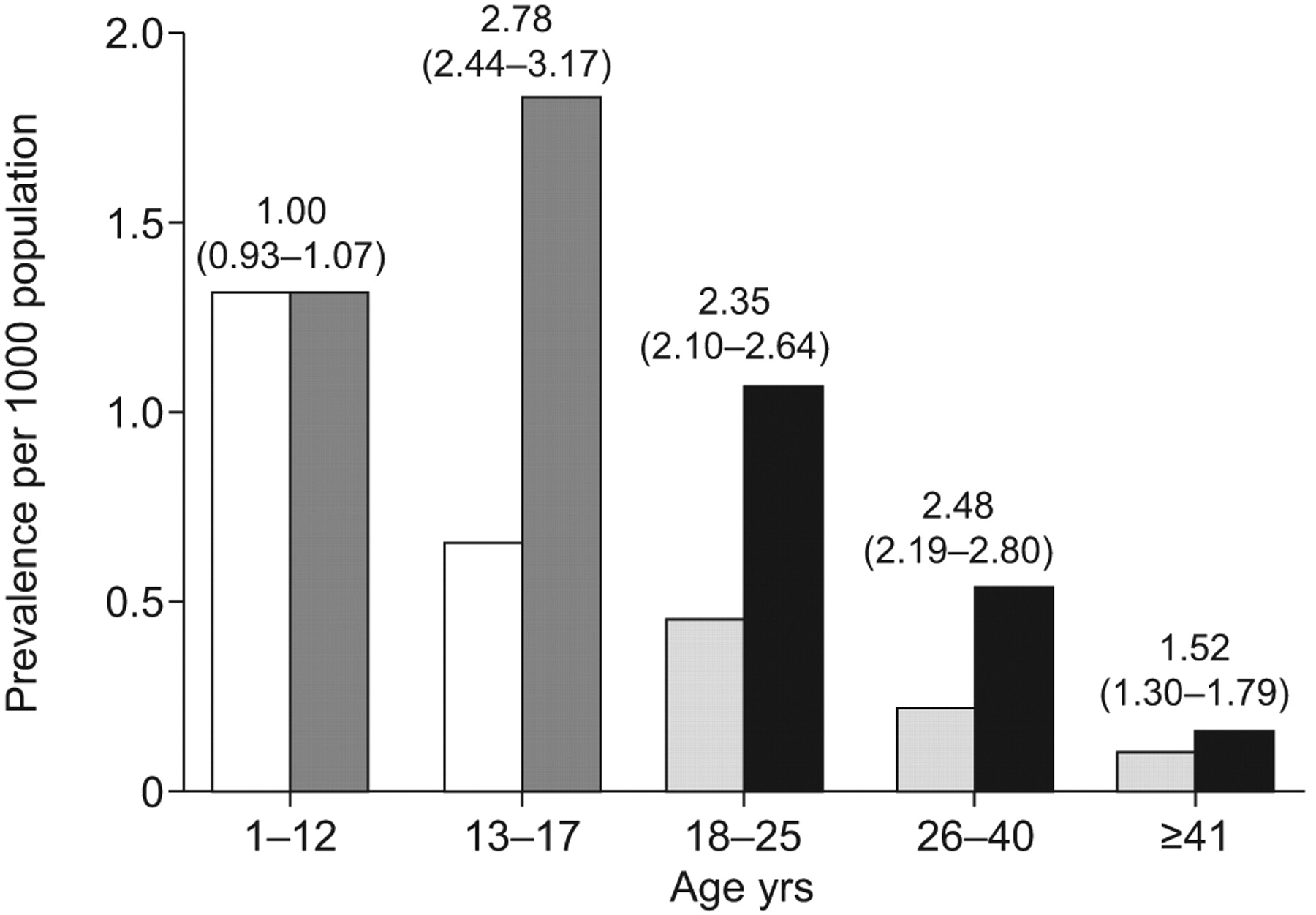
Comparison of prevalence of congenital heart disease in 1985 and 2000 between patients of different age groups. □: children, year 1985; ▒: children, year 2000; ░: adults, year 1985; ▪: adults, year 2000. Prevalence ratios (95% CIs) for year 2000 versus 1985 are also shown. Reproduced from [9] with permission from the publisher.
In adults and children, PAH-CHD is essentially the same disease, although there may be differences between the two age groups reflecting the natural history of PAH. PAH-CHD is best considered an ongoing, progressive disease, as some patients may present with less (albeit increasingly) severe PAH-CHD at an advanced age. The pathophysiology is mostly similar, although in the paediatric population there are anomalous cases where the PAH is apparently not causally related to the CHD. Such cases are not explained by the physiology of the cardiac defect (e.g. the presence of small, haemodynamically insignificant shunts), are discordant with the expected natural history of the defect, or resemble idiopathic PAH in the setting of CHD. Examples include patients with small atrial septal defects or ventricular septal defects, right pulmonary artery arising from the aorta, transposition of the great arteries and pulmonary vein stenosis, or, more rarely, patients with Scimitar Syndrome.
Idiopathic PAH has seen substantial research interest in recent years. While data from patients with idiopathic PAH cannot be directly extrapolated to the end-stage PAH-CHD population, the two conditions share similar lung histology. Compared with idiopathic PAH, however, adults with PAH-CHD have a better prognosis [11]. Progression of idiopathic PAH is rapid and survival may be limited to a few years after diagnosis, while progression of PAH-CHD is slower and patients may survive for decades after diagnosis [12]. One study reported 77% survival at 3 yrs for PAH-CHD versus 35% for idiopathic PAH [13].
PATHOPHYSIOLOGY AND NATURAL HISTORY
A wide range of cardiac defects can be associated with PAH (table 1⇓), and the natural history of PAH-CHD varies according to the underlying defect.
Classification of lesions in congenital heart disease
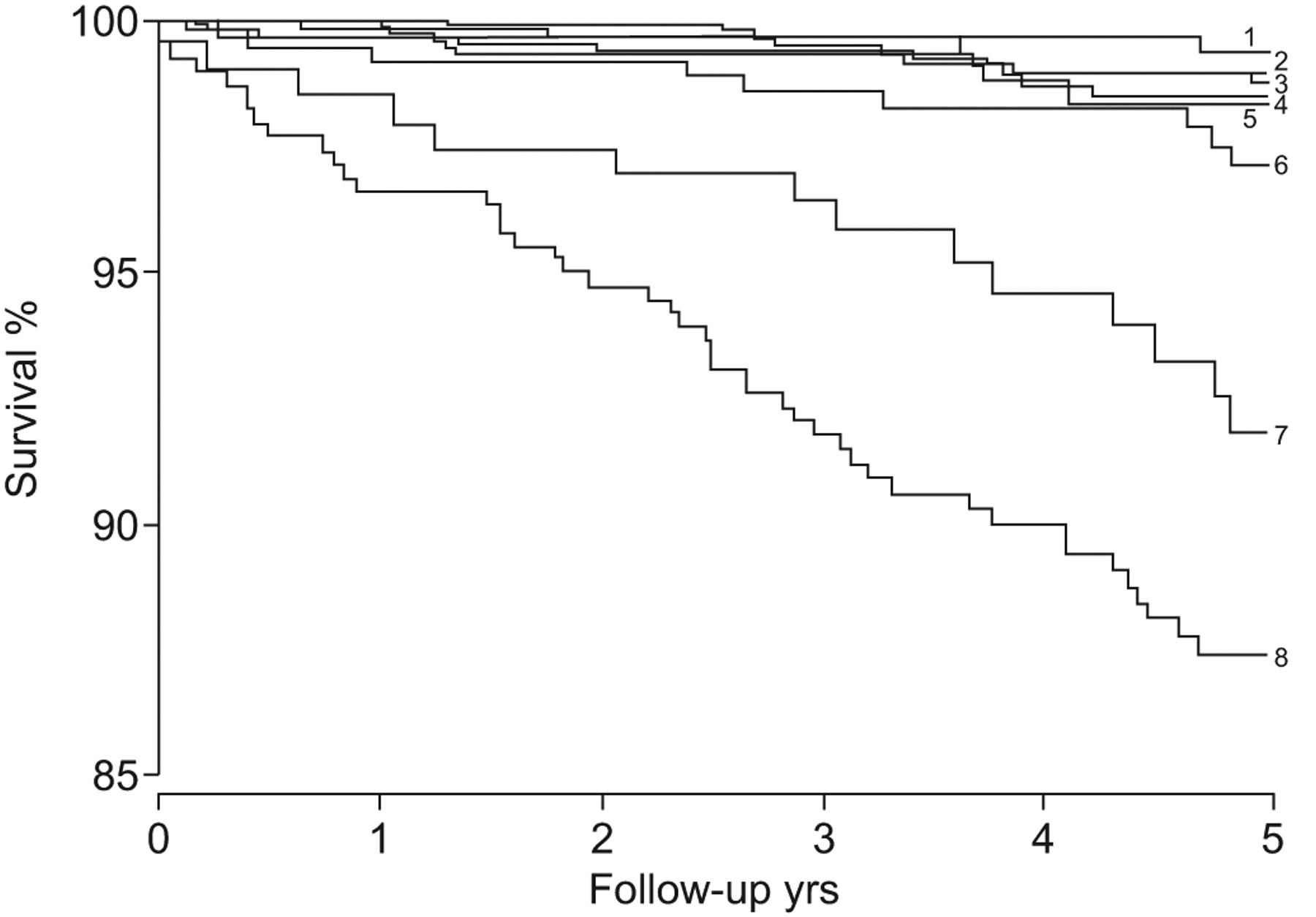
The likelihood of developing PAH also depends on the size and location of the defect. Even with a large atrial septal defect (simple CHD, pre-tricuspid location), severe forms of PAH are rare, while the incidence is 15–20% in patients with a sinus venosus defect, 50–70% with a large ventricular septal defect or a large patent ductus arteriosus (post-tricuspid location), and nearly 100% for adults with truncus arteriosus (complex CHD). The degree of compromise is higher [14, 15] and survival is lower with the more complex cardiac defects (fig. 2⇓) [16].
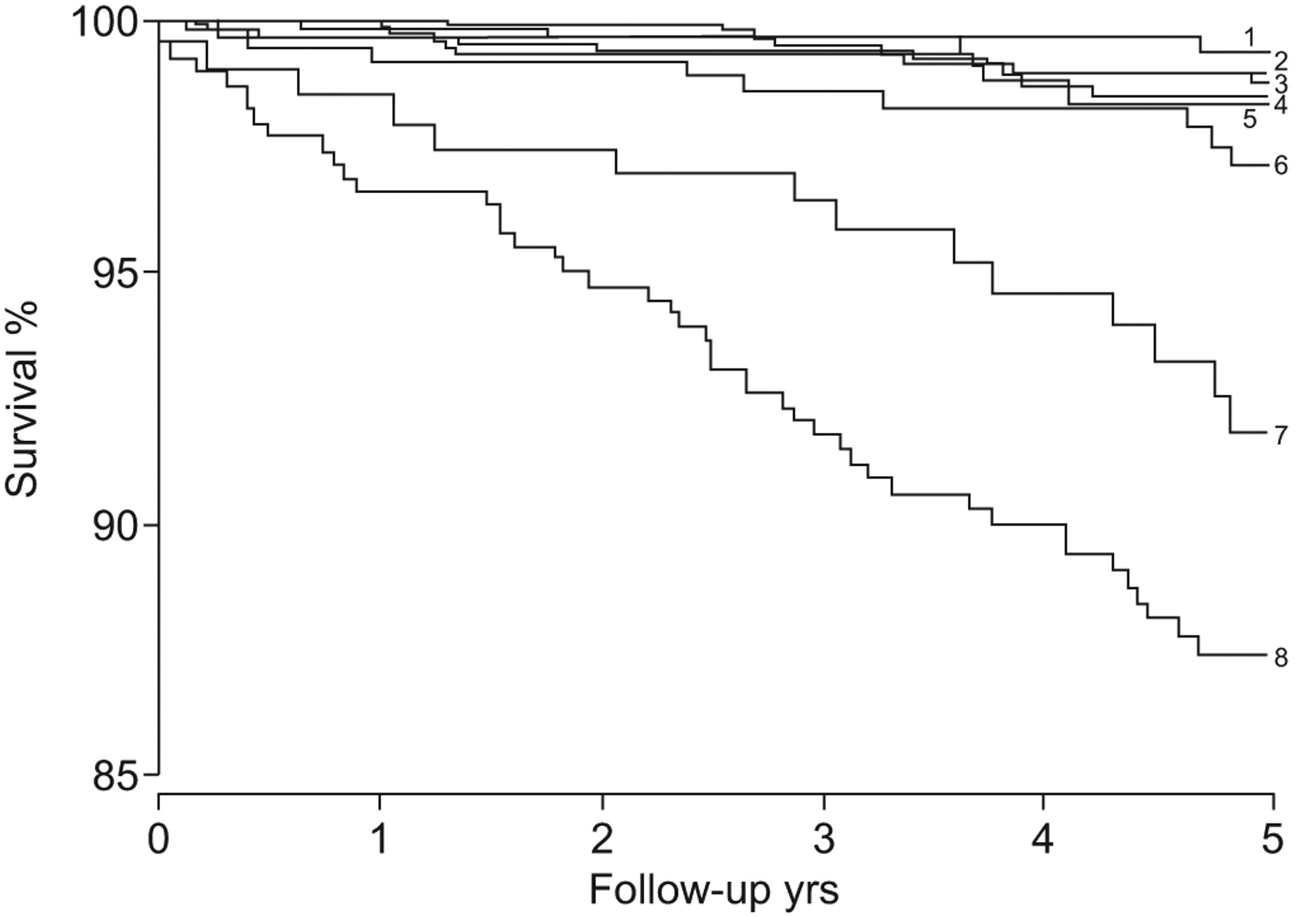
Survival in congenital heart disease is reduced among patients with more complex cardiac defects. 5-yr survival for each cardiac defect are as follows (greatest first, top to bottom on the figure): 1) coarctation of aorta 99.3%; 2) atrial septal defect 98.9%; 3) tetralogy of Fallot 98.7%; 4) ventricular septal defect 98.4%; 5) Marfan 98.3%; 6) transposition of the great arteries 97.1%; 7) Fontan 91.8%; and 8) cyanotic defect 87.4%. Reproduced from [16] with permission from the publisher.
The pathophysiology of PAH-CHD is, to some extent, similar to that of idiopathic PAH [10, 17, 18], both being due to vascular remodelling and increased pulmonary vascular resistance, although differences do exist. Cardiac defects resulting in left-to-right shunting cause PAH because the increased pulmonary blood flow induces increases in shear stress and circumferential stretch. These haemodynamic forces within the pulmonary vessels induce endothelial dysfunction [10]. This loss of endothelial barrier function may be associated with degradation of the extracellular matrix and the release of growth factors which, in turn, induce smooth muscle hypertrophy and proliferation, with migration into the pulmonary vasculature [10]. The vascular remodelling seen in PAH-CHD results in a progressive increase in pulmonary vascular resistance. The early lesions display medial hypertrophy (grade 1), progressing to intimal proliferation (grade 2), lumen occlusion (grade 3), progressive arterial dilatation and plexiform lesions (grade 4), thinning and fibrosis of the media (grade 5) and, finally, necrosis (grade 6) [19].
Once the increasing pulmonary vascular resistance equals systemic resistance, the left-to-right shunt becomes bi-directional, and once pulmonary vascular resistance exceeds systemic resistance, shunt reversal and the development of Eisenmenger syndrome occurs. Eisenmenger syndrome is the most advanced form of PAH-CHD and is characterised by bi-directional or right-to-left shunting and chronic cyanosis [18].
Cyanosis is common in patients with PAH-CHD. Right-to-left shunting reduces arterial oxygen capacity, resulting in hypoxic damage and multi-organ disease [10]. All patients with Eisenmenger syndrome have cyanosis, which is often severe, even at rest [12, 18]. This is commonly accompanied by secondary erythrocytosis, whereby an increased number of red blood cells are produced in an attempt to compensate for the hypoxia; if these are decreased (e.g. by venesection), the patient is at risk of becoming microcytic (a risk factor for cerebrovascular events), iron deprived and hypochromic. In contrast, cyanosis is seen in idiopathic PAH only when cardiac output is low, is rarely severe at rest, and is only rarely accompanied by (mostly uncharacterised) haematological manifestations [12]. The cyanotic complications seen in patients with Eisenmenger syndrome include ischaemic and embolic events (e.g. stroke), bleeding (e.g. haemoptysis) and thrombotic diathesis, intrapulmonary thrombosis, haematological involvement (e.g. erythrocytosis), infection (e.g. cerebral abscess), progressive right ventricular failure, cardiac arrhythmia and sudden cardiac death, renal or hepatic dysfunction, acne and skeletal disease (e.g. scoliosis) [10, 18, 19].
In contrast to idiopathic PAH, haemodynamics are typically more favourable in Eisenmenger syndrome [18]; cardiac output is preserved by the right-to-left shunting [12]. Mortality is, therefore, generally higher in idiopathic PAH compared with Eisenmenger syndrome. However, the cyanotic complications of Eisenmenger syndrome are associated with significant mortality [20]. In an Italian cohort of 188 patients with Eisenmenger syndrome followed for a median 31 yrs, the 61 deaths were largely attributed to sudden death (29.5%), heart failure (22.9%) or haemoptysis (11.4%) [21]. Those patients with Eisenmenger syndrome with a simple cardiac defect have significantly better survival than those with complex defects (fig. 3⇓) [21, 22]. The development of Eisenmenger syndrome in a patient with complex CHD is associated with a 10- to 12-fold increase in mortality [23].

Mortality in Eisenmenger syndrome is related to the underlying cardiac defect. #: p = 0.02, comparison between Eisenmenger patients with simple and complex lesions. Reproduced from [22] with permission from the publisher.
CLASSIFICATION AND DIAGNOSIS
PAH-CHD has been classified as belonging to the same broad PAH category as idiopathic PAH, familial PAH and PAH associated with other pathologies, including connective tissue diseases, infection and drugs [24]. With its inherent heterogeneity, PAH-CHD may warrant separate consideration [25]. A new classification system has been proposed, giving specific consideration to the type of cardiac diagnosis, the prognosis and evolution of pulmonary vascular disease, and the circulatory physiology (table 2⇓) [25]. This would be of value because in terms of management strategies, generalisation may be hazardous for such a heterogeneous disease [25].
Progress in the classification of pulmonary arterial hypertension associated with congenital heart disease
Several groups of patients are now recognised clinically. The first are patients with Eisenmenger syndrome, for whom treatment options are emerging. The second group are patients with a systemic-to-pulmonary shunt with increased pulmonary vascular resistance; their cardiac defect cannot be closed without high risks, and management options are currently limited. The third group of patients are those with small cardiac defects, which may not be the underlying cause of increased pulmonary artery pressure, only a related risk factor. The final group are patients who present with high pulmonary vascular resistance and high pulmonary artery pressure after corrective cardiac surgery, despite their normalised heart architecture [27]. These different groups require separate management strategies, and have different responses to treatment.
Clinical signs of PAH-CHD may include central cyanosis, clubbing, peripheral oedema, abdominal tenderness, right ventricular heave, a loud pulmonary ejection click and an accentuated pulmonary component of the second heart sound [10]. Murmurs associated with valvular regurgitation may be present, but murmurs due to previous left-to-right shunts at the ventricular or arterial level usually disappear upon development of Eisenmenger syndrome [10]. The diagnosis of PAH-CHD involves a thorough investigation using a variety of techniques. This should include radiography, ECG, measurement of arterial oxygen saturation and exercise tolerance, echocardiography with magnetic resonance imaging or computed tomography for detection and characterisation, and right heart catheterisation to confirm PAH and perform a complete haemodynamic study, including pulmonary vascular resistance measurements (table 3⇓).
Diagnostic tools in pulmonary arterial hypertension associated with congenital heart disease
MANAGEMENT
At the present time, management comprises the prevention of PAH in patients with CHD (by surgical repair of the shunt prior to the onset of pulmonary vascular disease), and the treatment of existing PAH (by surgery or medical management). The only curative option for end-stage disease is heart transplant surgery. Optimal timing of each therapeutic strategy is of great importance. More data are required, however, in order to determine the best time to initiate pharmacological or surgical intervention, as well as questioning whether patients should be treated, the groups of patients who should be treated, and which outcome measures should be used to monitor their response.
Surgical management
There are limited data with which to define higher “safe” values when considering surgical repair, but a threshold pulmonary vascular resistance of <6 Wood units after vasoreactivity testing is generally the consensus of opinion. Patients with values in the “grey zone” between 6–10 Wood units remain a particular challenge. In general, patients with left-to-right shunts with high pulmonary blood flow and low pulmonary vascular resistance are most likely to be operable. These patients may have atrial septal defects, ventricular septal defects or complex defects, with increased pulmonary blood flow index and pulmonary artery pressure, and are symptomatic with congestive heart failure. In contrast, among patients with bi-directional flow with normal or slightly increased pulmonary blood flow and moderately increased pulmonary vascular resistance, the chances of surgical reversal of PAH are low, and there is less consensus on optimal management. With shunt reversal and the development of Eisenmenger syndrome, surgery is contraindicated; at this point the lesions are considered irreversible.
The timing of surgery is critical, and progression of the disease and the onset of right ventricular failure and death are possible, despite successful surgical repair [28]. Surgery must be performed prior to the onset of high pulmonary vascular resistance. At this stage, early changes may be reversible after correction of the cardiac defect. If surgery is delayed, it is less effective. Correction of a ventricular septal defect at age ≤6 months results in normal pulmonary vascular resistance after 12 months; however, while delaying surgery until age ≥2 yrs results in a reduction in resistance, normalisation is not achieved [28]. It is, therefore, prudent that surgery should be very early in children with a massively increased blood flow [29]. However, these guidelines do not cover all eventualities. There are cases of presentation later in childhood (e.g. at age 8–10 yrs) with operable lesions, and also cases of 6-month-old babies presenting with severe cyanosis and high pulmonary vascular resistance.
The prognostic factors for PAH reversibility require identification, and there is a need for noninvasive markers. A study assessing the cellular and histological basis of reversibility of PAH-CHD found a strong association of irreversibility with impaired endothelial cell apoptosis and anti-apoptotic signalling from perivascular inflammatory cells [30]. Subsequently, the presence of high numbers of circulating endothelial cells in peripheral blood was identified as a potential biomarker for irreversibility in a comparison of patients with reversible (n = 16, median age 2 yrs) or irreversible (n = 10, median age 9 yrs) PAH, and controls (n = 5) [31]. There was no difference in the number of endothelial progenitor cells between the groups [31], although a previous study found decreased numbers in patients with Eisenmenger syndrome and idiopathic PAH compared with controls [32].
Medical management
For patients who are inoperable, especially those with Eisenmenger syndrome, the understanding of medical management is advancing.
Generally, all patients with PAH-CHD are advised to avoid dehydration, heavy physical exertion, and extreme isometric exercise, although low-grade aerobic exercise is recommended. Pregnancy [33] and high altitudes are to be avoided, although travel on commercial airlines can be undertaken [34]. Surgery can be undertaken with specific anaesthetic management, although care needs to be taken with angiography and noncardiac procedures. Supportive therapies include oral anticoagulant treatment in patients with pulmonary artery thrombosis and absent or mild haemoptysis, along with diuretics and management of iron deficiency. Supplemental oxygen may be useful [35], although the benefits of nocturnal delivery are controversial [36].
The medical management of PAH involves disease-targeted therapy, i.e. strategies aimed at the pathological mechanisms underlying PAH. There have been numerous controlled trials to optimise this for idiopathic PAH. However, in PAH-CHD, the evidence base for the various therapies is in its early stages; for most, there have been no dedicated controlled trials in PAH-CHD.
Prostacyclin synthase is reduced in patients with PAH, resulting in inadequate production of prostacyclin I2 (a vasodilator with antiproliferative effects), and the prostacyclin analogues, epoprostenol, treprostinil and iloprost, have been a traditional mainstay of the treatment of idiopathic PAH. There are few data for PAH-CHD, but the benefits appear to be similar. In an uncontrolled study of 20 children with PAH-CHD (mean age 15 yrs), 1 yr of prostacyclin therapy improved haemodynamics and quality of life [37], while in a retrospective observational study, severely impaired and symptomatic patients with Eisenmenger syndrome (n = 8, median age 37 yrs) had significantly improved functional capacity, oxygen saturation, and haemodynamics following 3 months' epoprostenol therapy [38]. In a mixed population of 39 children with PAH of various aetiologies (including patients with PAH-CHD), epoprostenol improved survival (84% at 3 yrs), functional status, exercise tolerance and ability to thrive [39]. However, the intravenous delivery of these drugs is a drawback, both practically and owing to the risk of infection. Among 39 children, 38% had catheter-associated problems, with 43 prescriptions for antibiotics, and 0.33 Hickman line changes per patient, per year [39].
Phosphodiesterase type-5 (PDE-5) inhibitors, such as sildenafil and tadalafil, inhibit the degradation of PDE-5, the enzyme responsible for hydrolysing the vasodilatory cyclic guanosine monophosphate. These compounds enable vasodilation in PAH, although there are limited data on their efficacy for PAH-CHD. A 12-month, open-label study of children with PAH (n = 14, of whom 10 exhibited PAH-CHD) reported improvements in exercise capacity and haemodynamics with sildenafil [40]. Similarly, a 6-month, prospective, open-label trial of sildenafil therapy found a significant reduction in systolic and mean pulmonary artery pressures and pulmonary vascular resistance, and improved cyanosis and functional capacity, in patients with Eisenmenger syndrome (n = 7) [41]. Similar benefits were seen in 16 patients with Eisenmenger syndrome after 12 weeks' therapy with tadalafil [42].
Endothelin is a mediator that drives fibrosis, vascular hypertrophy, proliferation and vasoconstriction, and, therefore, underlies the pathophysiology of PAH and correlates with disease severity and outcome [43, 44]. Consequently, endothelin receptor antagonists, such as bosentan, sitaxentan and ambrisentan, have been a focus of research for PAH. Of these, the dual endothelin receptor antagonist bosentan has strongest supportive dataset of all targeted therapies for PAH-CHD. A prospective, open-label study of 21 patients with PAH-CHD (including 15 with Eisenmenger syndrome) reported that 16 weeks' treatment with bosentan resulted in clinical, exercise, and haemodynamic improvements [45]. Similarly, in an open-label, prospective, multicentre study, adults with PAH-CHD (n = 33, of whom 23 had Eisenmenger syndrome) showed improvements in functional status and exercise capacity after bosentan treatment for a mean of 2.1 yrs [46]. These findings were replicated in the only randomised controlled trial dedicated to PAH-CHD. The Bosentan Randomised Trial of Endothelin Antagonist Therapy-5 (BREATHE-5) was a 16-week, double-blind and placebo-controlled trial of bosentan in patients with Eisenmenger syndrome in New York Heart Association functional class III (n = 54) [47]. Compared with placebo, bosentan significantly reduced pulmonary vascular resistance (-472.0 dyn·s·cm−5; p = 0.038) and mean pulmonary arterial pressure (-5.5 mmHg; p = 0.036), and significantly increased 6-min walk distance (+53.1 m; p = 0.008), without compromising peripheral oxygen saturation (fig. 4⇓) [47]. These findings were sustained in the open-label extension [48], and a subgroup analysis showed that the improvements were irrespective of the underlying cardiac defect [49].

Significant improvements in a) pulmonary vascular resistance (placebo group, n = 17; bosentan group, n = 36) and b) exercise capacity (placebo group, n = 17; bosentan group, n = 37) in patients with Eisenmenger syndrome treated with bosentan. PVR: pulmonary vascular resistance; 6MWD: 6-min walk distance. #: decrease of 472.0 dyn·s·cm−5, p = 0.038; ¶: increase of 53.1 m, p = 0.008. Reproduced from [47] with permission from the publisher.
Recently, a new approach to the treatment of PAH-CHD has been proposed. This involves treat-and-repair, whereby a patient previously considered irreversible (for example with Eisenmenger syndrome) is first treated with targeted therapy to reduce their PAH, before undergoing surgery to repair the cardiac defect [50]. More data are needed to determine the long-term benefits and risks of this approach.
Transplantation
Transplantation surgery, either by heart/lung transplant or a lung transplant plus corrective cardiac surgery, is the only potentially curative option for PAH-CHD. This approach is, however, not without limitations. The 10-yr survival for a transplanted heart/lung is around 30–40%, which is low compared with the expected survival of patients with Eisenmenger syndrome, making it difficult to determine optimum timing for transplant. The need for transplant might, however, be delayed by the use of targeted therapies. A retrospective study of 43 patients with Eisenmenger syndrome found that the mean time to death or inscription on the active transplant waiting list was significantly longer for those treated with prostacyclin analogues or endothelin receptor antagonists (7.8 yrs) compared with those who did not receive targeted therapy (3.4 yrs; p = 0.006) [51]. However, delaying the need for transplant may not be beneficial for a disease with slow progression; especially in the presence of any age restrictions for acceptance onto the transplant list. The criteria and prognostic indicators for transplant in this population are unclear and warrant consideration.
Management considerations
The consensus for optimal management of patients with Eisenmenger syndrome has recently been updated (fig. 5⇓) and its multidisciplinary nature is clear. The importance of patient education and engagement also cannot be over-emphasised, especially in terms of endocarditis prophylaxis, lifestyle issues (especially exercise, contraception and the risks of pregnancy), and frank discussion about future prospects and prognosis. Historically, there has been a lack of effective communication with patients, resulting in a failure of patients to engage in their own welfare, leading to large and unacceptable losses to follow-up. Other considerations include the increasing need to improve communication with adult (as opposed to paediatric) clinicians, to reflect the shift in demographics to a more adult population of patients with PAH-CHD.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Management algorithm for patients with Eisenmenger syndrome. PAH: pulmonary arterial hypertension; CHD: congenital heart disease; MRI: magnetic resonance imaging; CT: computed tomography; 6MWT: 6-min walk test; NYHA: New York Heart Association. Reproduced from [52] with permission from the publisher.
CONCLUSIONS
In conclusion, improvements in diagnosis and surgical and medical management have improved the long-term survival prospects for patients with PAH-CHD, resulting in an increase in the number of patients surviving to adulthood. Although there are some differences in aetiology, treatment response and survival, paediatric and adult patients with PAH-CHD have essentially the same disease; a complex condition with a natural history that varies depending on the underlying cardiac defect and multiorgan chronic adaptation to it. Benefits of novel targeted therapies in PAH-CHD are increasingly clear, although the less well characterised treatment options require further, long-term investigation in PAH-CHD populations. Of increasing interest is the management of patients with complex congenital defects, along with determination of the operability (and timing) of patients in order to optimise outcomes, each of which warrants further investigation.
Support statement
M.A. Gatzoulis has received support form the Clinical Research Committee at the Royal Brompton Hospital and British Heart Foundation, UK. R. Alonso-Gonzalez has received support from the Fundacion Alfonso Martin Escudero, Madrid, Spain.
Statement of interest
M.A. Gatzoulis has been on the advisory board of Actelion Pharmaceuticals, Pfizer and GlaxoSmithKline, and has received unrestrictive educational support from Actelion Pharmaceuticals and Pfizer. He has also acted as an expert witness on this subject. R. Alonso-Gonzalez has been a paid lecturer for Actelion and GlaxoSmithKline. M. Beghetti has served on advisory boards/consulted for Pfizer, Actelion Pharmaceuticals, Bayer Schering, Encysive, GlaxoSmithKline, INO Therapeutics, Eli Lilly and Mondobiotech; has received lecture fees from Actelion Pharmaceuticals, Encysive and Bayer Schering; and has received reimbursement for attending symposia from Bayer Schering and Actelion, and for organising their educational symposium once a year in Switzerland.
Provenance
Publication of this peer‐reviewed article was supported by Actelion Pharmaceuticals Ltd, Switzerland (unrestricted grant, European Respiratory Review issue 113).
Acknowledgments
The authors thank A. Gray from Elements Communications Ltd (Westerham, UK) for medical writing assistance supported by Actelion Pharmaceuticals (Allschwil, Switzerland).
- Received June 1, 2009.
- Accepted June 19, 2009.
- © ERSJ Ltd
References









