





Abstract
Insulin resistance is often associated with obstructive sleep apnoea syndrome (OSAS) and could contribute to cardiovascular risk in OSAS. Sleep loss and intermittent hypoxia could contribute to the pathogenesis of the metabolic alterations associated with obesity, a common feature of OSAS. The biology of the adipocyte is being increasingly studied, and it has been found that hypoxia negatively affects adipocyte function.
In November 2007, the European Respiratory Society and two EU COST Actions (Cardiovascular risk in OSAS (B26) and Adipose tissue and the metabolic syndrome (BM0602)), held a Research Seminar in Düsseldorf, Germany, to discuss the following: 1) the effects of hypoxia on glucose metabolism and adipocyte function; 2) the role of inflammatory activation in OSAS and obesity; 3) the alarming rates of obesity and OSAS in children; 4) the harmful effects of the metabolic syndrome in OSAS; 5) the effects of OSAS treatment on metabolic variables; and 6) the relationship between daytime sleepiness and hormonal and inflammatory responses. Insulin resistance in skeletal muscle, the role of the endocannabinoid system and novel pharmacological approaches to treat insulin resistance were also discussed.
As obesity and hypoxia could be the basic links between OSAS and adipocyte dysfunction, further research is needed to translate these new data into clinical practice.
The Research Seminar “Metabolic aspects of obstructive sleep apnoea syndrome” was organised by the European Respiratory Society (ERS) in close collaboration with two EU-funded COST (Cooperation in Scientific and Technological research) Networks: Cardiovascular risk in obstructive sleep apnoea syndrome (OSAS) (B26) and Adipose Tissue and the Metabolic Syndrome (BM0602). The COST programme aims at building pre-competitive research networks in Europe [1] and has been included in the 7th Framework Programme in recognition of its high scientific impact and cost-effectiveness. This Seminar was the first to be organised under the joint sponsorship of the ERS and COST, and the involvement of two COST Actions reflects the multidisciplinary and open exchange of ideas promoted by the COST programme among European scientists.
The topic of the Seminar was chosen based on the strong need felt by clinicians and basic scientists interested in OSAS to discuss adipose tissue biology with experts, in order to improve the approach to metabolic derangements in OSAS. OSAS is often associated with obesity in adults [2]. Clinicians dealing with OSAS patients need to gain a deeper insight into the pathophysiology of adipose tissue because prevalence of obesity is growing worldwide at an alarming rate [3]. Prevalence of obesity is also rising in children, and paediatric OSAS shows a phenotype shift, from a “local” to a “systemic” disease, with increasing similarities to adult OSAS [4]. Therefore, metabolic problems can be expected to become a major issue in the clinical management of OSAS at all ages.
The association of OSAS and metabolic dysfunction represents a harmful background, which may have profound cardiovascular consequences but is still incompletely understood. OSAS patients are at an increased risk for fatal and nonfatal cardiovascular events, and treatment with nasal continuous positive airway pressure (CPAP) is effective in decreasing such risk [5, 6], thus suggesting a causal relationship between OSAS and atherosclerosis [7, 8]. However, the complexity of OSAS pathophysiology makes it very difficult to assess the independent role of intermittent hypoxia (the main culprit), obesity and sleep disruption in the accelerated progression of atherosclerosis seen in OSAS patients. The precise mechanism(s) are still unclear, but an increasing number of studies suggest that OSAS, inflammatory activation and metabolic disturbances are closely linked [9].
During the past 10 yrs, the view that adipose tissue is mainly a depot organ has profoundly evolved. The adipocyte is now considered as a major actor in metabolic and inflammatory regulation [10]. Current research efforts from several laboratories throughout the world aim to define the molecular mechanisms underlying the metabolic alterations of obesity in order to develop more effective treatment strategies. Since the adipose tissue is a complex endocrine organ involved in the regulation of food intake, an inadequate amount of sleep or poor sleep quality may affect brain physiology and contribute to the pathogenesis of obesity, as recently suggested [11].
According to clinical and epidemiologic studies, the cluster of risk factors known as the metabolic syndrome (MetS) is associated with an increased risk for diabetes, cardiovascular events and mortality in the general population [12]. Although the definition of the MetS is still under debate [13, 14], insulin resistance (IR) is considered, by most studies, as the major metabolic abnormality and is usually associated with an increased amount of visceral (dysfunctional) fat [15]. The definition of the MetS by the World Health Organization is based on the direct measurement of IR [16]. The definition by the National Cholesterol Education Program-Adult Treatment Panel III is based on simple clinical findings (abdominal obesity, dyslipidaemia, hypertension and increased plasma glucose), and is easily applicable as it does not require tests to be performed in a specialist environment [17]. Finally, the definition proposed by the International Diabetes Federation shares many features with the Adult Treatment Panel III definition, but defines the cut-offs for waist circumference according to ethnicity [18], thus accounting for differences in body habitus between Caucasian and Asian populations. All these definitions should be considered as “in progress” and subject to change based on the evidence provided by current and future studies.
Irrespective of the definition chosen, the MetS is often found in OSAS patients from different locations worldwide [19–22]. However, the relative role played by OSAS and obesity in the pathogenesis of MetS remains uncertain. It is possible that OSAS and MetS exert a synergistic negative effect on metabolism, but little evidence is available at present. Most studies on IR in sleep-disordered breathing (SDB) were cross-sectional, and severe OSAS was usually associated with the highest level of obesity. However, IR was found in nonobese OSAS patients [23] and improved after OSAS treatment in some studies [24, 25].
Based on these findings, the purpose of the Seminar was to gather together experts on both OSAS and adipose tissue biology in order to discuss the current knowledge in both areas, identify potential areas of common interest for future research and possibly establish collaborative projects according to specific expertise and research interests in both areas. Due to this, care was taken to ensure the faculty and participants attending the seminar were balanced in terms of experience and knowledge, in order to foster multidisciplinary discussion among experts in both fields. The Seminar was organised into three sessions, each of which lasted for half a day with three to four lectures by faculty members. A summary of the meeting and conclusions are reported in the following sections.
SESSION 1. SLEEP APNOEA, OBESITY AND SLEEP DISRUPTION: MULTIPLE MECHANISMS AFFECTING METABOLISM
During this session, J. Montserrat (Barcelona, Spain) and F. Giorgino (Bari, Italy) introduced the topic of metabolic alterations in OSAS by analysing different aspects of sleep apnoea and their potential link with metabolic derangements.
M.R. Bonsignore (Palermo, Italy) provided an overview of the topic by briefly illustrating: 1) the most relevant clinical and pathophysiological features of OSAS; 2) the evidence for increased cardiovascular risk associated with OSAS; and 3) the effectiveness of long-term treatment with nasal CPAP in reducing the incidence of fatal and nonfatal cardiovascular events [5, 6]. Following this, the available data on metabolic disturbances in OSAS were reviewed.
Several studies have reported the association of OSAS with glucose intolerance and IR [19–24], but not all of them found an independent relationship between SDB and metabolic disturbances after correction for obesity [25–27]. A longitudinal study found an increased incidence of diabetes in patients with OSAS, but the significance of OSAS disappeared after accounting for obesity [28]. Moreover, OSAS treatment with CPAP did not affect insulin sensitivity or glycaemic control in randomised controlled studies in diabetic [29] and nondiabetic patients [30]. However, other data support the possibility that OSAS may favour the development of IR in the absence of obesity, since acute exposure to hypoxia caused IR in normal subjects [31], and lean mice exposed to intermittent hypoxia developed IR [32].
Although the effects of hypoxia on the adipose tissue are still incompletely defined, adipose tissue hypoxia was shown to occur in obesity [33, 34]. Adipocytes exposed to hypoxia in vitro produced less adiponectin (i.e. an important adipokine with protective effects against IR) compared with cells exposed to normoxia [35]. In adipocytes and macrophages exposed to hypoxia, the following findings have been reported: 1) inflammatory activation within 2 h [33]; 2) transcription of genes regulated by the hypoxia inducible factor (HIF)-1 after 4–8 h [33]; and 3) evidence of endoplasmic reticulum stress [34]. Therefore, hypoxia may be a pathophysiologically important link between OSAS and obesity but, to date, no study has assessed the response of adipocytes to intermittent hypoxia, which represents the main feature of OSAS.
Prevalence of the MetS is higher in OSAS patients than in the general population or in obese non-OSAS subjects [36]. Markers of OSAS severity, such as the apnoea/hypopnoea index or the degree of oxygen desaturation, correlated with the amount of visceral fat [37] and, according to one study, visceral fat decreased after CPAP treatment [38]. Other studies, however, found that prevalence of the MetS was unaffected by CPAP treatment for a few weeks [29, 30], suggesting that long-term data on large samples of subjects are needed before any conclusion on this point can be made.
IR is the main pathophysiological feature of the MetS, and abnormalities in insulin–glucose metabolism have been repeatedly shown in OSAS patients [19], especially when OSAS is severe. OSAS may increase the risk of developing type 2 diabetes but data on this topic are controversial, similar to the effects of CPAP treatment on IR. These uncertainties are not surprising. Severe OSAS is usually associated with severe obesity, which is a major cause of IR and an important confounder for the independent effects of OSAS on metabolic variables. Another point of discussion is whether OSAS patients show the same components of the MetS as subjects without OSAS. This is an area under intense clinical investigation, together with OSAS-associated changes in adipokines, blood lipids and liver enzymes [9].
Sleep loss is an additional, potentially important mechanism by which OSAS may affect metabolism [39]. In healthy subjects, sleep restriction was associated with IR, increased appetite and craving for carbohydrates [40]. Short sleep duration was shown to be associated with altered levels of leptin and ghrelin, and increased body mass index (BMI) [41]. Finally, a prospective study reported that short sleep duration was a significant risk factor for the development of obesity in young adults [42], leading to speculation that the progressive fall in the time spent asleep over the past decades in the general population may have played some role in the increasing prevalence of obesity. A growing body of evidence indicates that not only total sleep deprivation, but also modest sleep loss may cause profound endocrinological changes. In young healthy subjects, glucose tolerance decreased after selective slow-wave sleep deprivation [43]. Partial sleep deprivation in otherwise healthy subjects induced a pro-inflammatory state, with increased release of interleukin (IL)-6 [44, 45] and production of IL-6 and tumour necrosis factor (TNF)-α by circulating monocytes [46]. The pro-inflammatory effects of sleep restriction may be, at least partly, mediated by the activation of the sympathetic nervous system [47, 48]. Therefore, although sympathetic hyperactivity in OSAS patients is generally considered a major consequence of intermittent hypoxia, it could be potentiated by sleep disruption and contribute to inflammatory activation.
R. Tkacova (Kosice, Slovakia) presented clinical data which indicated that most subjects with OSAS showed a 10-yr moderate-to-high cardiovascular risk (>10%) [49]. The MetS was found in 66% of OSAS patients, and preliminary results suggest that metabolic variables can improve after CPAP treatment for 2 months. While these data indicate that OSAS might play some role in the pathogenesis of metabolic abnormalities, more data and long-term follow-up studies are needed to confirm the positive effect of OSAS treatment on metabolic variables.
Alternatively, OSAS could also be considered as a feature of the MetS phenotype, as was proposed by A. Vgontzas (Hershey, PA, USA) in the second lecture of the session. Sleep apnoea has been considered to be a “local abnormality” of the upper airway rather than part of a “systemic illness”, despite the early recognition of the association between obstructive sleep apnoea (OSA), obesity and cardiovascular problems [50]. In 1997, Vgontzas et al. [51] first reported elevated IL-6 and TNF-α levels in patients with excessive daytime sleepiness (EDS), which positively correlated with BMI. In subsequent studies, elevated IL-6, TNF-α and insulin levels were found in patients with OSAS independent of obesity, and the amount of visceral fat was the primary factor linked to sleep apnoea [52]. Females with polycystic ovary syndrome (PCOS), a condition associated with IR, also showed a high prevalence of OSAS and daytime sleepiness [53]. Other findings supporting the view that sleep apnoea and sleepiness in obese patients may be manifestations of the MetS include: 1) the association of obesity and EDS in obese patients without sleep apnoea [54]; 2) the independent association of PCOS and type 2 diabetes with EDS after controlling for OSA, obesity and age [53, 54]; 3) the increased prevalence of sleep apnoea in post-menopausal females, and the reduced risk for OSA in females treated with hormone replacement therapy [55]; and 4) the lack of effect of CPAP treatment on hypercytokinaemia and IR indices in obese OSAS patients (A.N. Vgontzas, Dept of Psychiatry, Penn State College of Medicine, Hershey, PA, USA; personal communication). Finally, in the US population (Third National Health and Nutrition Examination Survey), prevalence of abdominal obesity [56] and symptomatic sleep apnoea [2] appears to increase in parallel. Therefore, a bi-directional, feed forward, pernicious association may exist among OSAS, EDS, inflammation and IR in promoting atherosclerosis and cardiovascular disease.
The alarming worldwide increase in obesity in children and adolescents, associated with changes in food consumption and low level of exercise, was discussed by W. Kiess (Leipzig, Germany). An increasing rate of type 2 diabetes mellitus has been recorded in the USA in children and adolescents, and more recently in many countries around the world. There is a high hidden prevalence and exact data on the epidemiology of type 2 diabetes in European children are lacking [57]. In Germany only 70 patients aged <15 yrs have been identified in the systematic, nationwide “DPV” Diabetes Survey, while calculations suggest that >5,000 youths would currently meet the diagnostic criteria of type 2 diabetes in Germany. In Australasia, the prevalence of type 2 diabetes is reportedly high in some ethnic groups and tightly linked to the obesity epidemic. No uniform or evidence-based treatment strategy is available. Many groups use metformin, exercise and nutritional programmes as a comprehensive approach to treat type 2 diabetes in childhood and adolescence. The lack of epidemiological data and the strong need for effective treatment strategies point to the key role of preventive programmes against obesity, focusing on exercise training and reduction of sedentary behaviour, encouraging healthy nutrition and supporting general education programmes [58]. Insufficient sleep is also a potentially modifiable factor involved in the pathogenesis of childhood obesity, as suggested by several studies [59–61], similar to the relationship found between short-sleep duration and obesity in adults [39].
There is substantial comorbidity of type 2 diabetes and obesity in childhood and adolescence [58], with hypertension, dyslipidaemia and psychosocial problems being the most common sequelae of primary childhood obesity. OSA represents an additional clinically threatening comorbidity in obese children. S. Verhulst (Antwerp, Belgium) discussed the high prevalence of SDB in overweight and obese children [62]; however, severe obesity is often associated with central rather than obstructive apnoeas [62]. Metabolic disturbances associated with paediatric OSAS and obesity were significantly correlated with the severity of SDB but the effect was rather small [63], suggesting a predominant role of obesity over that played by OSAS in obese children. Nevertheless, in overweight and obese children, OSAS was associated with increased uric acid, a marker of hypoxia, independent of body weight [64]. Moreover, a high prevalence of fatty liver disease and IR has been recently reported in obese/overweight children with OSA, with significant improvement in the metabolic profile following OSA treatment [65]. Both these reports suggest that OSAS worsens metabolism in children. Finally, short-sleep duration has been shown to be associated with IR in obese children with or without OSAS [66].
In summary, the first session of the Seminar provided a comprehensive overview of clinical and epidemiological data on metabolic disturbances in OSAS and obesity in both adults and children. The data are still limited and some controversy exists among experts as to the independent role of OSAS in IR and the MetS. However, it is clear that many pathophysiological features of OSAS (such as intermittent hypoxaemia, poor sleep quality or sympathetic hyperactivity) may contribute to metabolic disruption and the associated increase in cardiovascular risk.
SESSION 2. ADIPOSE TISSUE IS METABOLICALLY ACTIVE
During this session, S. Sasson (Jerusalem, Israel) and J. Kopecky (Prague, Czech Republic) analysed the metabolic aspects of adipose tissue, paying special attention to inflammation, IR, cardiovascular consequences and new therapeutic targets.
P. Trayhurn (Liverpool, UK) presented an introduction to this topic by discussing: 1) the role of white adipose tissue as a major secretory organ; 2) the role of hypoxia in adipose tissue and hypoxia-related signalling pathways; 3) the identification of hypoxia-sensitive genes in adipocytes; and 4) the implications of hypoxia for obesity-associated disorders. These topics have been addressed in recent reviews [67, 68].
It is now generally accepted that white adipose tissue is a key endocrine and secretory organ which releases a large number of adipokines with a major link to inflammation and immunity. The paradigm shift in adipose tissue biology was initiated in 1994 by the discovery of leptin [69]. Subsequently, a continually increasing range of proteins, peptides and other factors, collectively termed adipokines, has been described to be released from white adipocytes [70]. The majority of these adipokines are linked to inflammation and their production is increased in obesity. To date, the only known exception is adiponectin, which exerts anti-inflammatory and anti-diabetic activity and is reduced in obesity and type 2 diabetes. Adipose tissue inflammation, which is believed to be related to the enlargement of adipose tissue and the concomitant infiltration of macrophages, is thought to play a key role in the development of the MetS, type 2 diabetes and cardiovascular disease [71].
In 2004, Trayhurn and Wood [72] suggested that the initiation of adipose tissue inflammation may represent a specific response to relative hypoxia in clusters of adipocytes that become distant from the vasculature as cell size increases. Two very recent studies have demonstrated that hypoxia occurs in adipose tissue of obese mouse models [33, 34]. HIF-1 is considered the master regulator of oxygen homeostasis and plays a key role in the response to hypoxia in most tissues. Transcription factors, such as nuclear factor (NF)-κB and cAMP response element binding (CREB), are considered to be downstream targets of HIF-1. The number of known hypoxia-sensitive genes is continuously growing and, to date, >70 genes have been described as targets of HIF-1. These genes include proteins involved in angiogenesis, cell proliferation, apoptosis and energy metabolism [73]. Cell culture experiments have shown that hypoxia may increase both expression and secretion of a variety of inflammation-related adipokines, such as IL-6, macrophage migration inhibitory factor and vascular endothelial growth factor (VEGF). It can be concluded that hypoxia affects the function of adipocytes and promotes adipose tissue inflammation. This may play a critical role in obesity-related disorders and may trigger the development of peripheral IR and thus promote the development of type 2 diabetes and the MetS.
The impact of the adipocyte–myocyte axis for the induction of skeletal muscle IR in the context of the MetS was discussed by J. Eckel (Duesseldorf, Germany). In recent years, evidence has been presented that adipose tissue communicates with skeletal muscle and other organs not only via free fatty acids but also through various secretory products collectively designated as adipokines [74]. The direct crosstalk between human adipocytes and myocytes was first demonstrated in a co-culture model where both cell types were kept in the same medium [75]. Co-culture with adipocytes clearly leads to IR in skeletal muscle cells, as the latter display decreased insulin receptor substrate (IRS)-1 and Akt phosphorylation with subsequent inhibition of glucose transporter-4 translocation and glucose uptake. This experimental model has been extremely helpful in studying critical features of IR observed in skeletal muscle from obese and diabetic patients. Analysis of the secretion from primary human adipocytes revealed that these cells release classical adipokines, such as TNF-α, IL-6, leptin and adiponectin, and newly discovered adipokines, such as tissue inhibitor of metalloproteinases-1 and monocyte chemotactic protein (MCP)-1 [76]. Among the novel adipokines, MCP-1 was first described as a secretory product of monocytes and endothelial cells with a prominent role in arteriosclerosis but it is also associated with the obese state. MCP-1 exhibits IR-inducing capability in adipocytes and myocytes [67].
Increased expression and secretion of adipokines in obesity may be a marker of low-grade chronic inflammation in adipose tissue. Protein kinase C and inhibitory κB kinase (IKK) are two kinases known to be involved in inflammatory processes underlying IR. IKK influences insulin sensitivity, especially in skeletal muscle, through two different mechanisms. First, it can directly inhibit insulin signalling by phosphorylating IRS-1 on serine residues and, secondly, IKK activates NF-κB. In turn, NF-κB regulates production of pro-inflammatory cytokines such as TNF-α and IL-6 [77]. Accordingly, inhibition of IKK reduces the secretion of pro-inflammatory adipokines and prevents IR in vitro and in vivo.
The endocannabinoid system plays an important role in the regulation of appetite and is involved in the pathophysiology of weight gain [78]. A novel, still not well described, pathway may involve the cannabinoid receptor type 1 (CB1) in skeletal muscle. Adipocytes produce endogenous ligands for the CB1 receptor and human skeletal muscle cells express CB1 receptors at a high level. Furthermore, CB1 receptor blockers, such as rimonabant, are able to prevent the negative crosstalk between fat and muscle. More work will be required to define the precise role of CB1 signalling in the induction of IR in human skeletal muscle.
During this session, B. Staels (Lille, France) analysed the relationship between inflammation and vascular dysfunction, with special attention to circadian rhythms and the role of nuclear receptors. It is known that myocardial infarction, stroke and sudden cardiac death mostly occur in the morning, possibly in relation to sympathetic activation at awakening [79], while sleep appears as a generally protective state. Clock genes in the suprachiasmatic nucleus are the major determinant of circadian rhythm in humans, but circadian modulation has been recently described in peripheral tissues as well, including the vascular compartment [80]. These data suggest the intriguing possibility that sleep disorders may affect the cardiovascular system by several mechanisms, including disruption of cardiovascular circadian modulation. Moreover, metabolism and hormone release also show circadian rhythm [81], indicating a complex regulation by direct, indirect and behavioural influences over 24 h. Finally, mice carrying mutations in the Clock gene develop obesity and the metabolic syndrome [82], whereas mice lacking nocturnin, another component of the circadian pathway, are resistant to diet-induced obesity [83]. These observations suggest a major role of circadian modulation in energy balance. Testing the independent role of sleep and circadian variations on metabolism and cardiovascular function will require specifically designed studies.
Peroxisome proliferator-activated receptors (PPAR) are nuclear receptors involved in several metabolic and vascular abnormalities found in obesity, type 2 diabetes and atherosclerosis [84–87]. Three PPAR types have been described (α, γ and β/δ) with different expression and function in several tissues [84–87], including the vascular compartment. In general, PPAR activation results in anti-inflammatory, insulin-sensitising and anti-dyslipidemic effects, and PPAR agonists are currently used in prevention and treatment of type 2 diabetes and atherosclerosis.
The macrophage is a potential target of treatment with PPAR-α and PPAR-γ agonists. PPAR-α activation positively affects cholesterol traffic in macrophages [88]. In mice fed on a high-fat diet, macrophages in the adipose tissue were found to be activated (M1 phenotype) compared to the predominant anti-inflammatory M2 phenotype found in lean mice [89]. Conversely, monocytes treated with PPAR-γ agonists were primed to become anti-inflammatory M2 macrophages [90]. Therefore, pharmacological modulation by PPAR agonists may positively affect several pathogenetic steps of obesity, type 2 diabetes and atherosclerosis. The role of sleep apnoea in such context is still unexplored, and deserves further study.
In the final lecture, on the biological specificity of visceral adipose tissue and therapeutic intervention, F. Giorgino (Bari, Italy) described the specific role of visceral adipose tissue in the metabolic abnormalities associated with obesity, and explained the molecular basis of the different reactivity of various adipose tissue depots. Furthermore, novel anti-obesity drugs that could potentially target visceral obesity were discussed.
Subcutaneous fat represents about two-thirds of total body fat; however, it is now evident that the distribution of body fat is more important than its total amount [91]. Visceral adiposity is strongly connected to IR and the MetS, and this has been attributed to a distinct biological property of adipocytes in this depot. Insulin signalling is more rapid in visceral fat, and visceral adipose tissue in general is much more active compared to subcutaneous fat [92]. Thus, visceral adipocytes exhibit intrinsic properties that are more harmful for target tissues compared to other fat depots.
In addition to diet and exercise, pharmacological agents may be required to facilitate weight loss and maintain a reduced body weight. CB1 receptor blockers, such as rimonabant, are known to induce weight loss and improve a variety of cardio-metabolic parameters [78]. Furthermore, CB1 receptors have been found in peripheral organs including muscle and liver, suggesting that the clinical use of CB1 receptor blockers may improve the metabolic profile beyond the effects secondary to reduction in food intake and decreased body weight [78]. Results of the Rimonabant in Obesity (RiO) studies strongly suggested that CB1 receptor blockade may be a useful novel treatment for multiple cardio-metabolic risk factors in overweight and obese patients with a great potential to target visceral obesity [93–96]. Unfortunately, major side-effects of rimonabant have recently been reported.
In summary, the second session of the Seminar provided an overview on current basic and clinical research on adipose tissue and obesity. The complex picture of obesity may show peculiar aspects in patients with OSAS, due to occurrence of intermittent nocturnal hypoxia and profound sleep disturbances. More basic and clinical research is needed to understand the potential synergy of obesity and OSAS in increasing cardiovascular risk, and the pathways involved at different steps of atherogenesis. Similarly, very few data are available on the effects of new drugs for the treatment of obesity on the amount and quality of sleep. These aspects are important if these drugs are to be used in clinical practice.
SESSION 3. EXPERIMENTAL MODELS AND HUMAN STUDIES
The purpose of this session, chaired by J. Hedner (Goteborg, Sweden) and M. Simionescu (Bucharest, Romania), was to discuss some relevant data obtained in human and animal studies on the interactions between intermittent hypoxia, inflammation, daytime somnolence and metabolic alterations in OSAS.
W. McNicholas (Dublin, Ireland) discussed the role of intermittent hypoxia in sleep apnoea as a major cause of inflammatory activation. Inflammation is a critical step in the pathogenesis of atherosclerosis, and contributes to different stages of atheroma formation [97]. Increased levels of circulating inflammatory markers, such as TNF-α, IL-6, IL-8 and C-reactive protein could be useful biomarkers to predict cardiovascular risk [98]. The same mediators [99–102] were found to be increased in untreated OSAS patients and significantly fell after effective CPAP therapy [102, 103], strongly supporting a pivotal role of inflammation in the accelerated progression of atherosclerosis seen in OSAS patients.
Cell culture and animal studies have improved our understanding of the underlying mechanisms of this association. Intermittent hypoxia, the hallmark of OSAS, was found to preferentially activate pro-inflammatory transcription factors, such as NF-κB and activator protein-1 [100, 104]. These factors activate various inflammatory cells, particularly lymphocytes and monocytes [105, 106], with the downstream consequence of expression of pro-inflammatory mediators that may lead to endothelial dysfunction [107]. Conversely, exposure to continuous hypoxia preferentially activated the transcription of genes dependent on the HIF-1 pathway, thus generating an “adaptive” response to hypoxia [100, 108]. HIF-1 dependent gene products (VEGF and erythropoietin) are little or not increased in OSAS patients, supporting a limited role of HIF-1 activation in the pathogenesis of OSAS-associated derangements [100, 101]. However, as the complexity of the HIF pathway is better understood, it is possible that HIF could play a role in mediating some of the effects of intermittent hypoxia, specifically acting at the level of the carotid body [109]. For example, HIF-1α-deficient mice did not develop systemic hypertension after chronic exposure to intermittent hypoxia [110], and a feed forward mechanism involving reactive oxygen species generation and HIF-1α during chronic intermittent hypoxia has been postulated [109]. Recent experimental data indicate that HIF-1 activation can be stimulated by NF-κB through reactive oxygen species production [111].
Besides the effects of OSAS on oxygenation, sleep deprivation may also contribute to systemic inflammatory activation [112]. Finally, inflammatory activation is a major feature of obesity [113], indicating that multiple causes are likely to be responsible for inflammation in OSAS patients. Nevertheless, the reported effects of CPAP treatment on inflammatory mediators seem to support an independent role of OSAS in causing systemic inflammation.
L. Lavie (Haifa, Israel) presented her recent studies on the biology of inflammatory cells in peripheral blood in OSAS patients. In particular, she discussed the response of circulating monocytes and neutrophils to intermittent hypoxia in OSAS patients, since both these cell types contribute to atherogenesis [97]. Intermittent hypoxia affects the production of radical oxygen species (ROS) and expression of adhesion molecules by monocytes and neutrophils [114]. OSAS patients showed increased ROS production and expression of selectins, CD15 by both neutrophils and monocytes, whereas expression of integrins CD11b and CD11c was only increased in monocytes. Moreover, monocytes adhered more avidly to endothelial cells in culture, while polymorphonuclear cells (PMNs) did not.
Neutrophils and monocytes may participate in different stages of atherogenesis by different mechanisms. In addition to activation, increased formation of ROS [115] and selectin-CD15 adhesion molecules, neutrophils obtained from OSAS patients showed decreased programmed cell death (apoptosis) [116], this in agreement with data indicating that hypoxia selectively inhibits constitutive neutrophil apoptosis [117]. Decreased apoptosis results in functional longevity of PMNs, therefore allowing for a higher potential for production of ROS, proteolytic enzymes and cytokines which facilitate endothelial cell damage. Monocytes also undergo activation, with increased formation of ROS, selectin CD15, integrins CD11b and CD11c, and adhesion molecules [114], but their pattern of activation indicates a predominant effect of increased adhesion to endothelial cells and transmigration into the vessel wall. Activation of lymphocytes γδ also occurs in OSAS [118], suggesting their possible intervention in the late stages of plaque formation [97]. Collectively, these data indicate that intermittent hypoxia plays an important role in OSAS-associated inflammatory cell activation.
A. Vgontzas (Hershey, PA, USA) challenged the belief that sleep apnoea and sleep disruption per se are the primary determinants of obesity-related daytime sleepiness and fatigue. Obesity is associated with objective and subjective EDS regardless of sleep apnoea and sleep loss in humans [119–121] and animals [122]. Indeed, within the morbidly obese, those who have high sleep efficiency at night are sleepier than those who have low sleep efficiency [123]. In addition, in large random samples of the general population, the primary determinants of subjective EDS were depression and metabolic disturbances, i.e. obesity/diabetes, and not sleep apnoea or objective sleep loss [54]. Sleepiness and fatigue are often found in conditions associated with IR, for instance PCOS, independently of sleep apnoea or obesity [124], or in conditions of insufficient physical activity [125]. Therefore, objective sleepiness may be primarily related to metabolic factors, whereas fatigue appears to be related to psychological distress [125].
The level of sleep/arousal within the 24-h cycle might be determined by the interaction of the hypothalamic-pituitary-adrenal (HPA) axis and pro-inflammatory cytokines, i.e. “hypercortisolaemia” plus hypercytokinaemia is often associated with low-sleep efficiency and fatigue, for example in subjects with insomnia [126], whereas “eucortisolaemia” or “hypocortisolaemia” plus hypercytokinaemia appears to be associated with high-sleep efficiency and objective sleepiness [127].
C. O'Donnell (Pittsburgh, PA, USA) discussed the development of IR after chronic exposure to intermittent hypoxia in a murine model [32]. This animal model allows the study of the effects of intermittent hypoxia on IR independent of any confounding effect of obesity. Indeed, clinical data are often equivocal, in part, because of the overwhelming confounding effects of obesity in both sleep apnoea and IR [19].
IR occurred during periods of intermittent hypoxic exposure in lean mice [32]. The increases in whole-body IR in the murine model were accompanied by a specific impairment of glucose uptake by oxidative muscle fibres. Periods of IR during exposure to intermittent hypoxia were associated with large increases in glucocorticoids. Furthermore, many of the “lipotoxic” mechanisms known to cause IR were also activated by exposure to intermittent hypoxia [32]. These data from a rodent model may provide insights to guide the focus of future clinical studies. Clearly, more translational studies are necessary to reconcile the data between animal models of exposure to intermittent hypoxia and the inconsistent therapeutic benefits of CPAP on IR in patient populations [29, 30].
There are several “classic” modulators of IR, such as elevated sympathetic nerve activity (SNA), the HPA and counter regulatory hormones that are potentially activated in patients with sleep apnoea. Although SNA is known to be elevated during both the daytime and night-time in sleep apnoea patients [128], the studies in mice indicated that an intact autonomic nervous system was not required for the development of IR. However, exposure to intermittent hypoxia caused significant increases in corticosterone levels, the predominant glucocorticoid in rodents. The role of cortisol in sleep apnoea as a potential mediator of IR has not been comprehensively examined.
More recently, the underlying mechanisms of obesity-related IR have focused on a stress/pro-inflammatory state characterised by hyperlipidaemia [10, 129], excess fat deposition [130], increased inflammatory cytokines [131], generation of reactive oxygen species, and activation of IKK/IκB/NF-κB and other pro-inflammatory pathways [113, 131]. There is also evidence in rodent models that intermittent hypoxia can worsen IR in obese mice [132], produce hyperlipidaemia, cause triglyceride accumulation and lipid peroxidation in the liver [133, 134], and activate the NF-κB pathway [105].
M. Dematteis (Grenoble, France) further discussed the role of acute sleep fragmentation and intermittent hypoxia in modulating glucose homeostasis in mice. Despite the existence of multiple plausible pathways for the hypoxic stress and sleep disruption of sleep apnoea to induce IR, the human data remains equivocal regarding the presence of causality. The overwhelming impact of obesity on both IR and sleep apnoea in the clinical setting have obscured clear insights in this emerging area of research.
In summary, the last session of the Seminar provided an insight into the molecular mechanisms involved in the pathogenesis of metabolic and inflammatory changes after exposure to intermittent hypoxia. The data are still limited, but point to a physiologically important interaction between hypoxia and metabolism, possibly involving the autonomic nervous system, oxygen radical production, and multiple pathways (NF-κB, HIF and apoptosis) and cell types (inflammatory cells, vascular endothelium and adipocytes).
CONCLUSIONS
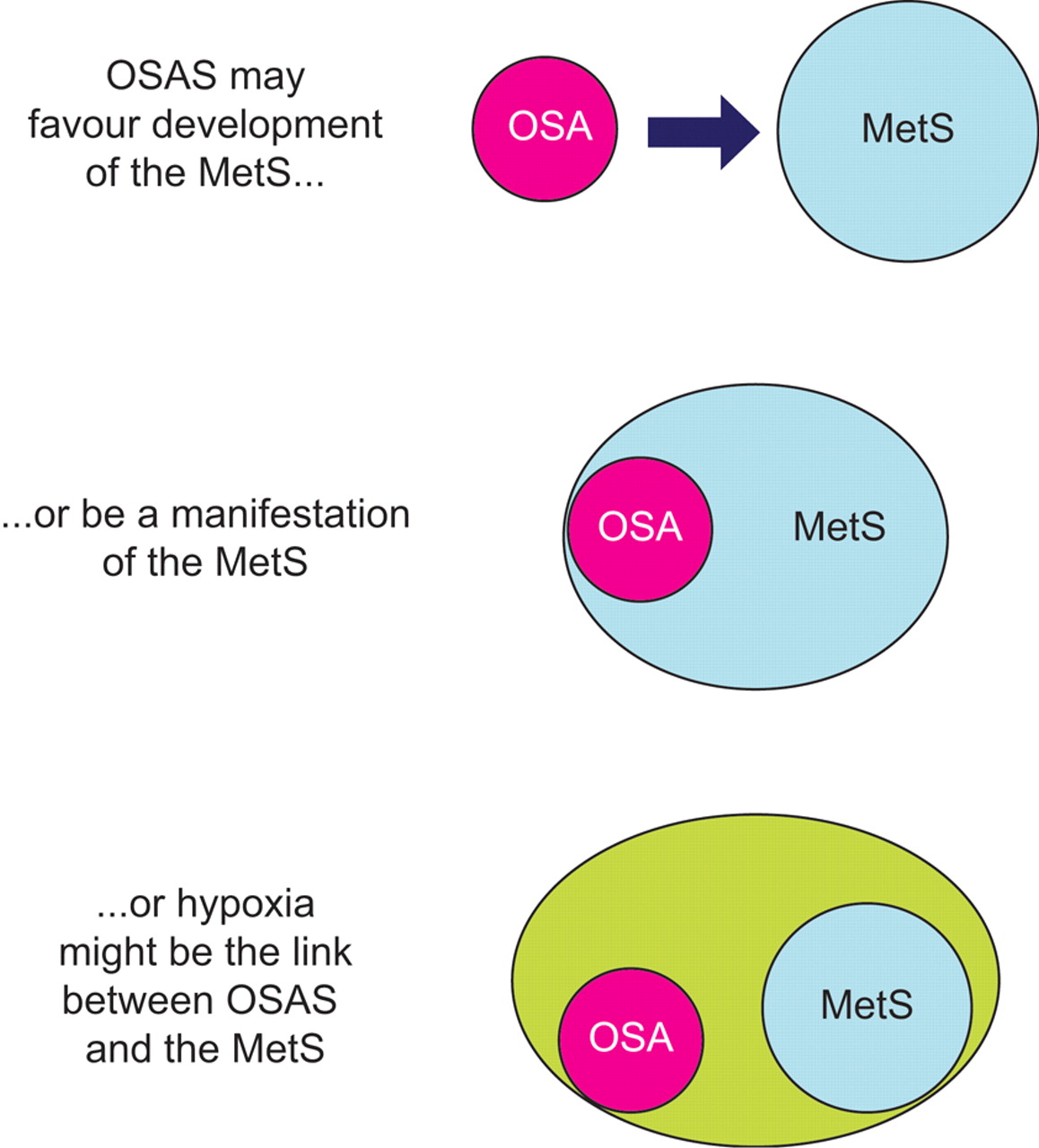
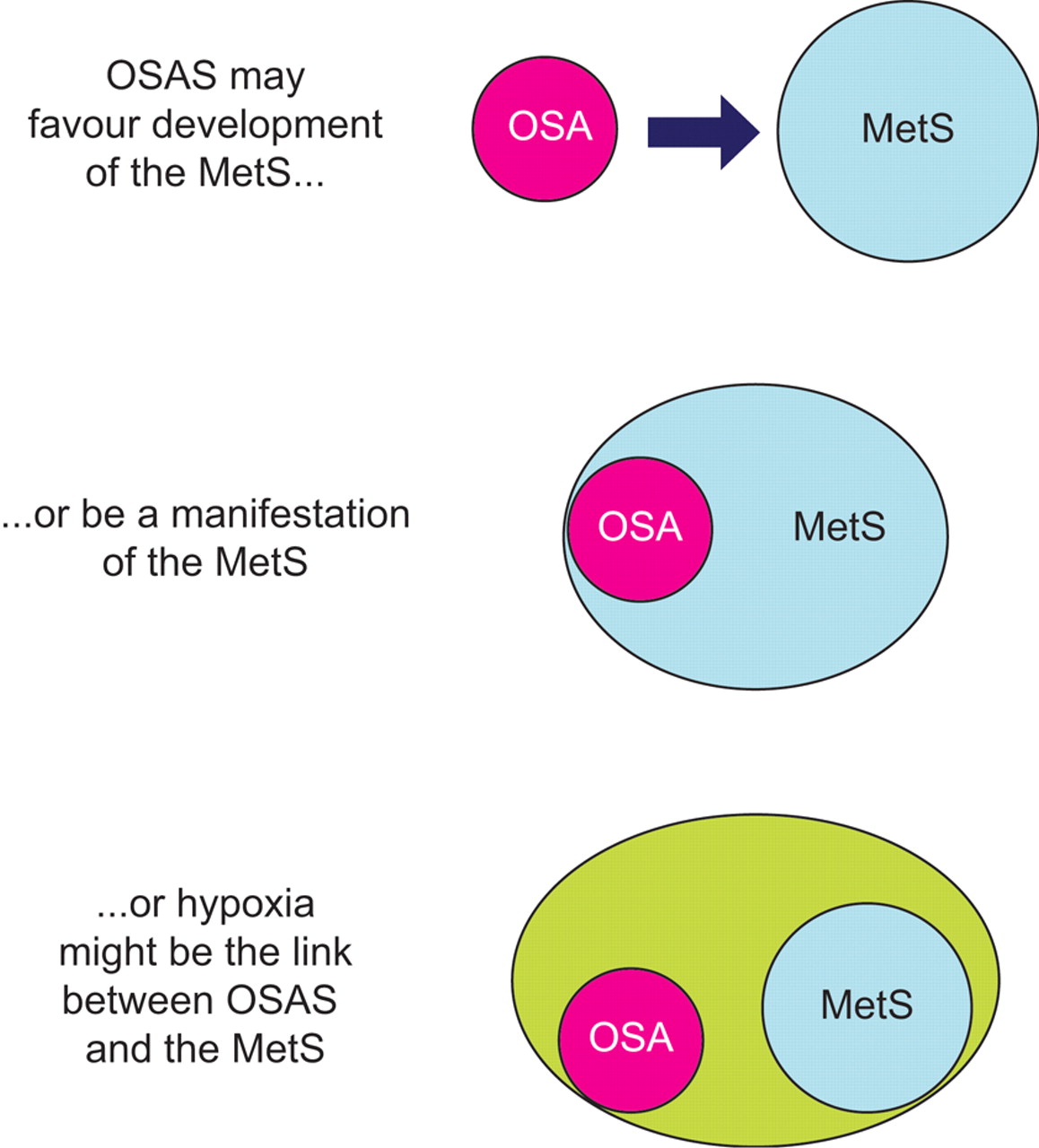
Figure 1⇓] schematically depicts the main theories on the relationship between OSAS and metabolic derangements, as discussed in the previous sections. Although it is premature to draw conclusions, all three possibilities are supported by clinical and experimental observations, and can be considered as starting points for further research.
{kind=link}
Schematic diagram of the current theories linking obstructive sleep apnoea syndrome (OSAS) and the metabolic syndrome (MetS). For more details refer to the text. OSA: obstructive sleep apnoea.
A number of questions were raised during the Seminar which deserve to be addressed in future studies. The most important one is the need to consider the frequent occurrence of OSAS and its potential contribution to clinical disease in obese subjects. While pulmonary physicians have been dealing for years with the multidisciplinary problems associated with SDB, the importance of OSAS is still relatively underestimated by clinicians looking after obese patients. A change in such attitudes would provide better studies on clinical series of overweight/obese patients.
From the biological point of view, there is a need to further characterise the adipocyte response to hypoxia, in particular the dose–response relationship. We do not know whether a critical level or duration of hypoxic stimulation is necessary to evoke a response by the adipocyte, whether subcutaneous and visceral adipocytes differ in their response to hypoxic exposure, and the possible differences in response to intermittent versus continuous hypoxia. The macrophage plays an important role in atherosclerosis and in obesity, suggesting potential benefits of pharmacological modulation of the M1 and M2 phenotypes in cardiovascular prevention in patients with either condition. Again, the effects of intermittent hypoxia on these functional features of the macrophage are largely unknown.
The available evidence suggests that CPAP quickly improves OSAS-associated pro-inflammatory changes, while exerting a slow or no effect on metabolic changes. The reasons for such difference are not understood at present, but may be revealed by future long-term studies exploring both metabolism and inflammation in OSAS patients.
The data on the relationship between hypoxia and adipocyte metabolism raise questions on the effects of hypoxia on metabolism in respiratory failure in general. Nutritional alterations in chronic obstructive pulmonary disease (COPD) are a major research topic, but the contribution of hypoxia and inflammation to adipose tissue dysfunction in COPD is unknown. The effect of hypoxia on in vitro adipocytes opens an entirely new field of investigation that extends well beyond the boundaries of SDB.
The possibility that poor-quality sleep affects IR opens the way for studies on the role of the central nervous system and hormonal changes in energy metabolism. The recent experimental data suggesting profound interactions between circadian regulation and energy homeostasis open a new point of view in the already complex field of sleep and metabolism. Many adipokines have central and peripheral effects, but the role of sleep deprivation or poor sleep quality in their modulation is still largely unknown.
The ultimate goal of the Seminar was the clinical translation of basic research to improve care of OSAS patients, help them lose weight and decrease their cardiovascular risk. This task can now begin to be addressed, similar to the efforts of the cardiologists who have been successful in identifying several drugs for primary and secondary prevention of ischaemic heart disease. Clinical research tailored to the specific issues raised by OSAS and obesity is urgently needed, especially as their prevalence in the population is constantly rising.
Statement of interest
None declared.
Provenance
The article is a summary of the ERS Research Seminar, which was held in Düsseldorf, Germany, on November 30 to December 1, 2007. Submitted article, peer reviewed.
Acknowledgments
The faculty members of the Research Seminar were as follows. M.R Bonsignore (Chair; Palermo, Italy), J. Eckel (Co-Chair and Chair of COST Action BM0602; Düsseldorf, Germany), W. Kiess (Leipzig, Germany), F. Giorgino (Bari, Italy), L. Lavie (Haifa, Israel), W. McNicholas (Chair of COST Action B26; Dublin, Ireland), C. O'Donnell (Pittsburg, PA, USA), B. Staels (Lille, France), P. Trayhurn (Liverpool, UK), A. Vgontzas (Hershey, PA, USA)
The participants of the Research Seminar were as follows. Iceland: E. Arnardottir and T. Gislason. Finland: M. Atalay and T. Saaresranta. Romania: R. Bercea, E. Ciomu, C. Guja, A. Herescu, D. Muntean, M. Simionescu and D. Todea. India: B. Bhushan. Spain: A. Campo Ezquibela, J. Duran, A. Martinez Valverde, C. Monasterio, J. Montserrat, and N. Salord. Switzerland: J-L. Carpentier and M. Niessen. Belgium: W. De Backer, J. Verbraecken and S. Verhulst. France: M. Dematteis. Poland: A. Dembinska-Kiec and J. Zielinski. Slovakia: V. Donic, D. Gasperikova, I. Klimes, M. Pallayova and R. Tkacova. Germany: J.K. Eckardt, S. Famulla, S. Grether-Beck, D. Lamers, K. Nehrenheim, R. Schulz, H. Sell, J. Swifka, A. Taube and D. Tews. Sweden: J. Hedner and M. Ryden. Norway: J. Jensen. UK: A. Jones, R. Riha and J. Steier. Israel: N. Kaiser and S. Sasson. Czech Republic: J. Kopecky and M. Pretl. Serbia: I. Kopitovic. Greece: I. Koutsourelakis and P. Steiropoulos. Slovenia: M. Kreftand and R. Zorec. Italy: C. Lombardi, O. Marrone, G. Parati, O. Resta and A. Zito. Turkey: F. Ozcan. The Netherlands: E. Rutten. Austria: A. Valipour. Lithuania: G. Varoneckas. Latvia: A. Vitols.
The authors would like to thank N. Vogel (European Respiratory Society), and K. Kostelidou and A. Van Der Snickt (COST) for excellent and dedicated assistance in the organisation of the Seminar. The authors also thank and A. Zito and O. Marrone for help in writing and revising the manuscript.
- Received January 14, 2009.
- Accepted January 26, 2009.
- © ERSJ Ltd
References









