





Abstract
Endothelin is one of a number of profibrotic cytokines and growth factors, along with transforming growth factor-β, connective tissue growth factor and tumour necrosis factor-α, thought to be involved in the pathogenesis of idiopathic pulmonary fibrosis (IPF) and other interstitial lung diseases characterised by fibrosis, including IPF-related pulmonary hypertension.
A growing body of evidence has supported a mitogenic effect of endothelin on fibroblasts and demonstrated that endothelin can reduce collagen breakdown and induce the synthesis of extracellular matrix components, all contributing to fibrosis. These findings, together with the detection of elevated levels of endothelin in the plasma and bronchoalveolar lavage fluid of patients, provide a sound rationale for dual endothelin receptor antagonism as a potential therapy for IPF and other fibrotic lung diseases.
The randomised controlled trial of Bosentan Use in Interstitial Lung Disease (BUILD)-1 investigated the potential of bosentan for the treatment of idiopathic pulmonary fibrosis. Although bosentan did not improve exercise capacity, encouraging trends were seen in the pre-defined clinically relevant end-point of disease progression or death, as well as dyspnoea and quality of life measures. The potential efficacy of bosentan, a well-established pathogenic mediator in pulmonary arterial hypertension, will continue to be evaluated in the treatment of idiopathic pulmonary fibrosis.
- Bosentan
- dual endothelin receptor antagonism
- fibrosis
- idiopathic pulmonary fibrosis
- interstitial lung disease
Diffuse (interstitial) lung disease (ILD) is a term that encompasses a heterogeneous group of lung conditions (fig. 1⇓) characterised by lung injury, a variable parenchymal immune/inflammatory response and, frequently, fibrosis of the interstitium. Taken together, these result in loss of lung function and often a poor prognosis. ILD may occur as a result of environmental or occupational factors (such as asbestos exposure), as a complication of connective tissue diseases (such as systemic sclerosis (SSc; scleroderma) [1]), or be of unknown aetiology (as is the case for the idiopathic interstitial pneumonias, including their most malignant variant, idiopathic pulmonary fibrosis (IPF)). On histopathology, IPF is characterised by the patchy, heterogeneous, peripheral honeycombing, fibrosis and fibroblastic foci that are the cardinal features of usual interstitial pneumonia (UIP).
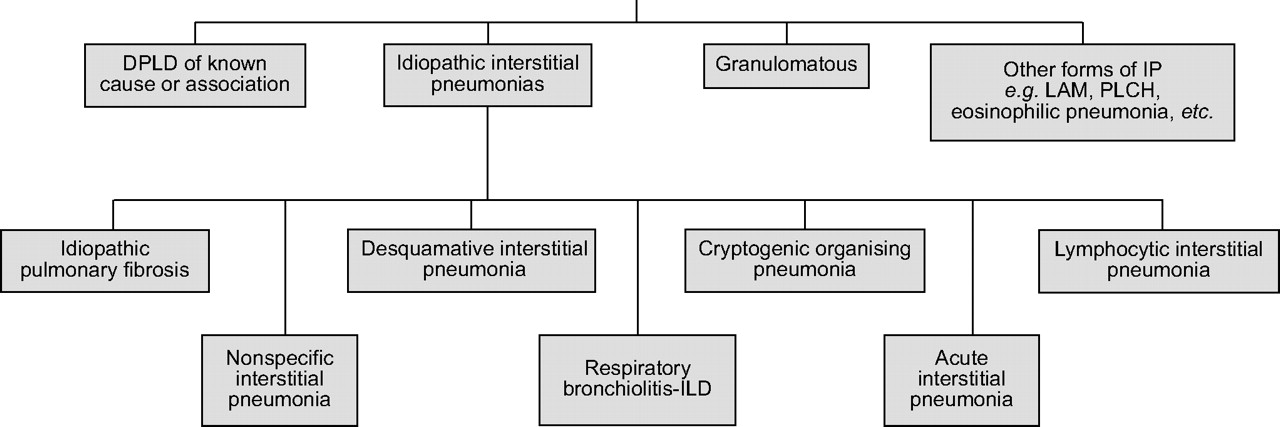
Classification of diffuse lung diseases. DPLD: diffuse parenchymal lung disease; IP: interstitial pneumonia; LAM: lymphangioleiomyomatosis; PLCH: pulmonary Langerhans’ cell histiocytosis; ILD: interstitial lung disease.
IPF is the most common form of ILD, the estimated annual incidence being approximately seven cases per 100,000 for females and 10 cases per 100,000 for males [2], although this is probably an underestimation. The prognosis of IPF is generally poor, with a median survival of <3 yrs [3, 4]. Symptoms of IPF include dyspnoea on exertion, nonproductive cough and inspiratory crackles heard on auscultation. Unfortunately, as is the case with pulmonary hypertension (PH), as a result of the subtle and nonspecific nature of early symptoms, patients typically present relatively late in the course of disease and the mean duration of symptoms prior to diagnosis is ∼2 yrs [2].
The development of PH can complicate many ILDs and is a common complication of IPF, having a negative impact on prognosis [5, 6]. In a recent study, PH was present in 31.6% of patients with advanced IPF undergoing assessment for transplantation, and was associated with a more than two-fold increased risk of death [5].
Although IPF was previously believed to be driven by a persistent and recurrent alveolar inflammatory response following alveolar injury, increasing evidence suggests that inflammation may play a less major role in its pathogenesis [2]. Current paradigms suggest that IPF may be the result of abnormal wound healing in response to alveolar injury (fig. 2⇓).
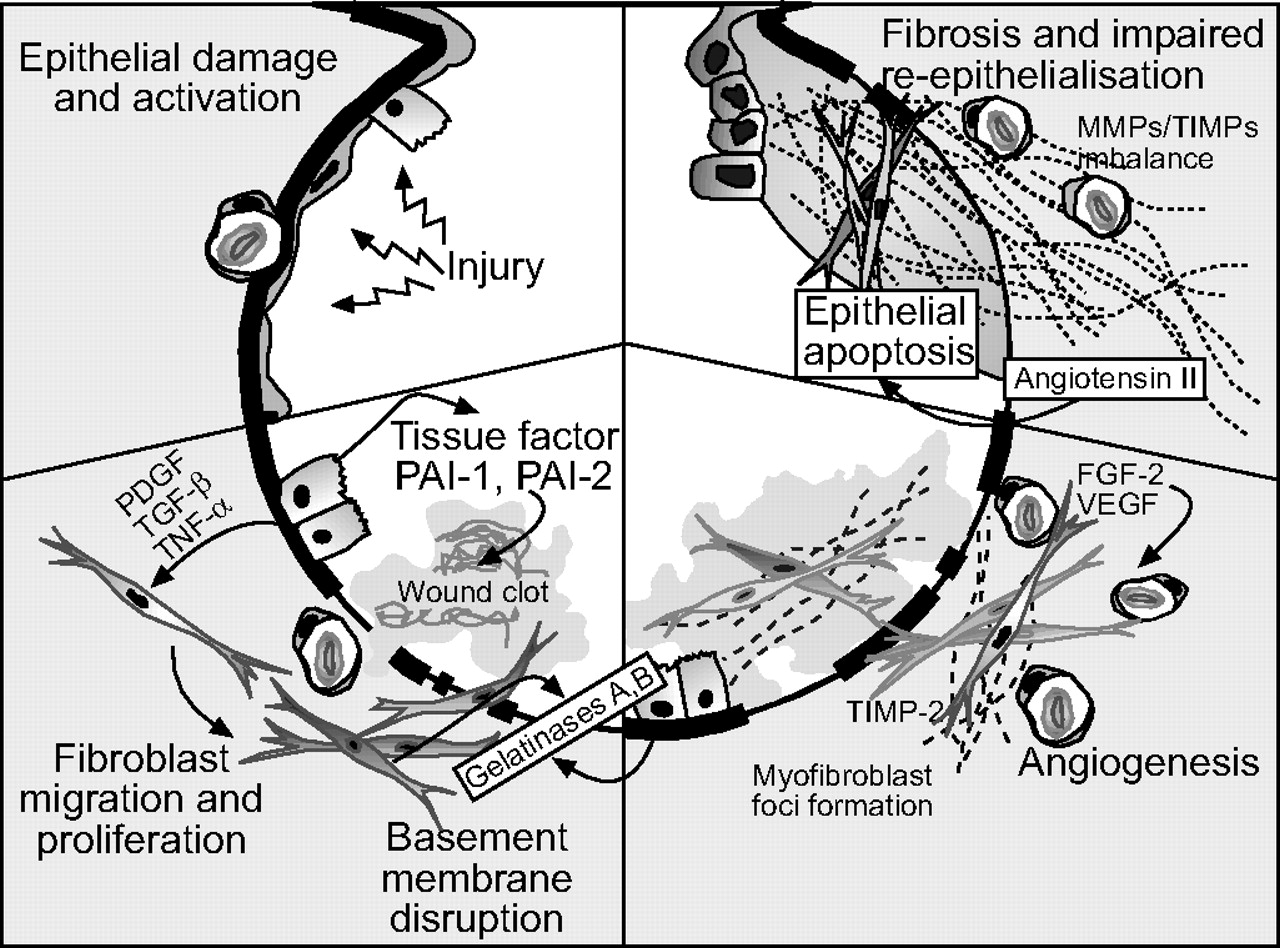
Pathogenesis of idiopathic pulmonary fibrosis (IPF). Multiple microinjuries damage and activate alveolar epithelial cells (top left), which in turn induce an antifibrinolytic environment in the alveolar spaces, enhancing wound clot formation. Alveolar epithelial cells secrete growth factors and induce migration and proliferation of fibroblasts and differentiation into myofibroblasts. Subepithelial myofibroblasts and alveolar epithelial cells produce gelatinases that may increase basement membrane disruption and allow fibroblast–myofibroblast migration (bottom left). Angiogenic factors induce neovascularisation (bottom right). Both intra-alveolar and interstitial myofibroblasts secrete extracellular matrix proteins, mainly collagens. An imbalance between interstitial collagenases and tissue inhibitors of matrix metalloproteinases (MMPs) provokes the progressive deposit of extracellular matrix (top right). Signals responsible for myofibroblast apoptosis seem to be absent or delayed in usual interstitial pneumonia, increasing cell survival. Myofibroblasts produce angiotensinogen, which, as angiotensin II, provokes alveolar epithelial cell death, further impairing re-epithelialisation. PDGF: platelet-derived growth factor; TGF: transforming growth factor; TNF: tumour necrosis factor; PAI: plasminogen activator inhibitor; TIMP: tissue inhibitors of metalloproteinases; VEGF: vascular endothelial growth factor; FGF: fibroblast growth factor. Reproduced and modified from [7] with permission from the publisher.
After initial alveolar injury resulting in epithelial cell damage, the wound-healing response is initiated to restore the integrity of the epithelial barrier. A key step in this process is rapid re-epithelialisation of the compromised area through epithelial cell migration, proliferation and differentiation. In IPF, this process appears to be slow and inadequate. The normal wound-healing process involves fibroblast migration, proliferation and differentiation into myofibroblasts and extracellular matrix (ECM) deposition. However, whereas during normal wound healing myofibroblasts apoptose and ECM is partially resorbed allowing epithelial cell migration and re-epithelialisation and complete tissue repair, in IPF these processes do not occur and discrete myofibroblast foci form in the injured areas, ECM continues to accumulate, the apoptotic process is impaired and re-epithelialisation cannot take place. These are the key pathogenic mechanisms that underpin IPF [2]. As a result of these processes, gas exchange is impaired, with progressive deterioration of pulmonary function and worsening prognosis.
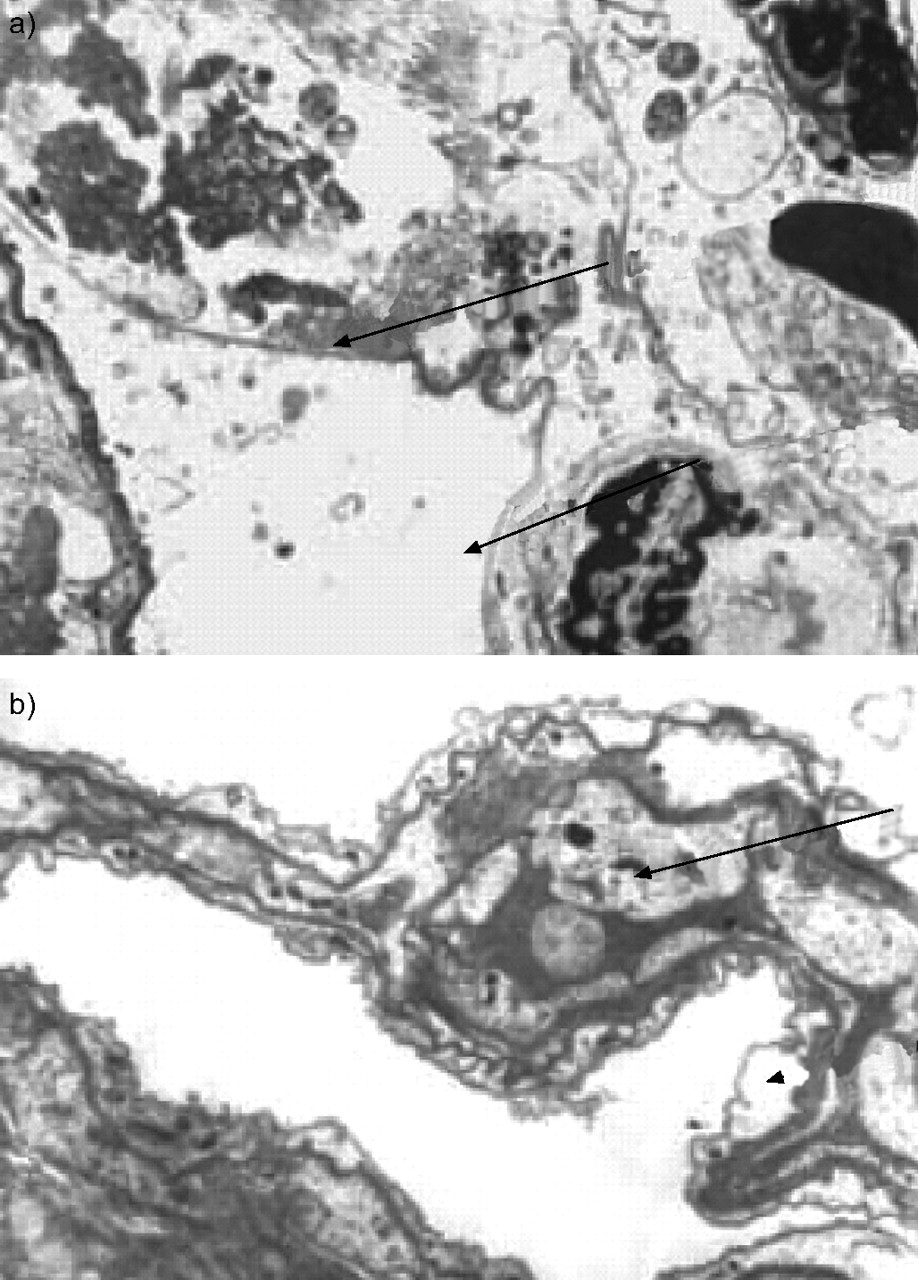
If alveolar injury resulting in epithelial cell triggering and subsequent aberrant wound healing is a critical and early stage in IPF, it should be possible to observe endothelial and epithelial cell damage in the absence of significant inflammation in the lung. Indeed, electron microscopic examination of a normal section of lung taken from a patient with SSc shows basement membrane that has lost its epithelium, as well as abnormal endothelial and epithelial cells, which may have been triggered or activated. These changes are observed in the absence of inflammation (fig. 3⇓) [8]. In IPF, the evidence is less strong but apoptosis has been shown in sections taken from biopsies of patients with IPF in areas of lung that were otherwise virtually normal on light microscopy, lending credence to the evolving epithelial/fibroblast paradigm of the pathogenesis of IPF [9].

Electron microscopy of a section of lung, normal on light microscopy, taken from a patient with systemic sclerosis. a) Arrows show bare basement membrane; b) the arrow shows the endothelial cell and the arrowhead shows the epithelial cell. Reproduced from [8] with permission from the publisher.
RATIONALE FOR USING ENDOTHELIN RECEPTOR ANTAGONISTS IN IPF
The triggering of an endothelial cell results in the upregulation and release of a number of pro-fibrotic cytokines and growth factors, including transforming growth factor (TGF)-β, connective tissue growth factor (CTGF), tumour necrosis factor (TNF)-α and endothelin (ET). All these factors are thought to be involved in the pathogenesis of IPF. Importantly, the close proximity of the epithelial and endothelial cells (fig. 3⇑) creates a microcompartment within the lungs, which includes epithelial cells that produce both ET and TGF-β and endothelial cells that can respond to TGF-β and produce ET.
There is now substantial evidence to support the notion that ET has mitogenic effects on fibroblasts, reduces collagen breakdown and induces the synthesis of ECM components, all contributing to fibrosis [10]. ET has been detected in the plasma of patients with IPF at significantly higher levels compared with matched controls (p<0.01) [11] and in patients with scleroderma, levels of ET in bronchoalveolar lavage fluid are higher in patients with lung fibrosis than in patients without lung involvement (fig. 4⇓) [12]. Importantly, biopsy studies have reported increased ET protein in the airway and alveolar epithelium of IPF patients, as well as in the vascular endothelium (fig. 5⇓) [11, 13].

Elevated levels of endothelin (ET)-1 in the plasma of a) idiopathic pulmonary fibrosis (IPF) patients and b) in the bronchoalveolar lavage fluid from scleroderma patients with and without lung fibrosis. SSc: systemic sclerosis; PF: pulmonary fibrosis. **: p<0.01; #: p<0.02. Reproduced and modified from [11] (a) and [12] (b) with permission from the publishers.

Endothelin (ET) protein is expressed in the bronchial and alveolar epithelium and also in vascular endothelium in idiopathic pulmonary fibrosis (IPF). ET-1 IR: ET-1-like immunoreactivity. ▒: IPF; ▓: IPF+pulmonary arterial hypertension; ░: nonspecific fibrosis. *: p<0.05 versus nonspecific fibrosis; **: p<0.01 versus nonspecific fibrosis. Reproduced and modified from [13] with permission from the publisher.
Moreover, ET has been found to stimulate pulmonary fibroblast proliferation [12] (fig. 6⇓) and the addition of ET to primary normal lung fibroblasts induces the expression of α-smooth muscle actin, which is the hallmark of the differentiation of fibroblasts to myofibroblasts, which in turn aggregate to form myofibroblast foci, which are characteristic features of UIP/IPF. In a transgenic mouse model, overexpression of ET results in the recruitment of inflammatory cells and the development of pulmonary fibrosis [14]. In vitro, ET induces myofibroblast formation and three-dimensional collagen lattice contraction [15].

Stimulation of pulmonary fibroblast proliferation by endothelin (ET). *: p<0.05; **: p<0.01. Reproduced from [12] with permission from the publisher.
The various profibrotic effects of ET demonstrated in vitro provide the rationale for the trial of endothelin receptor antagonists (ERAs) for ILDs characterised by extensive fibrosis and excessive ECM deposition due to the overproduction of collagen and the inactivation of collagenases. A number of experiments in vitro have supported the potential use of ERAs in the clinical setting. The dual ERA, bosentan, which blocks both the ETA and ETB receptors, has been shown to partially normalise ET-induced profibrotic changes characteristic of the scleroderma phenotype [15], particularly the fibrillar structure of α-smooth muscle actin, and to inhibit ET-induced three-dimensional collagen lattice contraction [15]. In addition, bosentan has been shown to decrease ET-induced collagen production in human fibroblasts [10] and to increase the levels of MMP-1, an enzyme that degrades collagen, back to normal (fig. 7⇓). Taken together, these in vitro experiments demonstrate that the use of bosentan favours a situation of reduced collagen deposition and all these effects of bosentan might be expected to be useful in the clinical management of patients with IPF.

Bosentan a) decreases endothelin (ET)-induced collagen formation in human fibroblasts and b) increases matrix metalloproteinase (MMP)-1 levels. *: p<0.05. Reproduced and modified from [10] with permission from the publisher.
CLINICAL APPLICATION OF BOSENTAN IN PATIENTS WITH IPF: THE BUILD PROGRAMME
Current treatment recommendations for IPF include anti-inflammatory agents (such as corticosteroids) and/or immunosuppressive/cytotoxic drugs (such as azathioprine), although the efficacy of these agents alone or in combination has not been demonstrated by clinical trials [7]. More recent studies have been relatively disappointing, although the use of N-acetylcysteine (NAC) as add-on therapy to prednisolone and azathioprine had a positive impact on lung function, slowing the deterioration of vital capacity and diffusion capacity relative to dual therapy and NAC placebo [16]. There is, however, an increasing need to attempt to modulate the fibrogenic component of IPF pathogenesis and a number of current approaches are targeting this pathway, including interferon-γ, pirfenidone and anti-TNF, as well as bosentan.
In light of the evidence discussed in support of ET as a key mediator in the pathophysiology of IPF-related PH in addition to IPF itself, it seems logical to suggest that the dual ERA bosentan may be predicted to provide clinical benefits in affected individuals. This was the rationale behind the BUILD (Bosentan Use in Interstitial Lung Disease) clinical study programme.
The randomised controlled trial BUILD-1 investigated the potential of bosentan for the treatment of IPF (fig. 8⇓). Although bosentan did not significantly improve the primary end-point measure, the 6-min walk distance (6MWD), encouraging trends were seen in the pre-defined end-points of combined incidence of disease progression or death at 12 months, representing a relative risk reduction of 38%. Improvements versus placebo were also observed in dyspnoea, as well as in measures of quality of life. Interestingly, in a subpopulation of patients with surgical lung biopsy-proven IPF, which represented two-thirds of the total patient population, many of these benefits were more pronounced, suggesting that to optimise therapy for patients, consideration may need to be given to subclassification of IPF. Importantly, the level of adverse events was consistent with that reported in randomised clinical trials of bosentan in pulmonary arterial hypertension [17, 18] and in everyday clinical practice [19].

BUILD (Bosentan Use in Interstitial Lung Disease)-1 study design.
In hindsight, the lack of observed effect on 6MWD may have been due, at least in part, to the inappropriateness of this measurement as an end-point in IPF. Although validated in the evaluation of treatment effects in PAH, the 6MWD is a novel, nonvalidated end-point for trials in IPF and may not be appropriate to measure disease progression or response to treatment in this patient population. However, the observed benefits with respect to the secondary end-points were sufficiently encouraging to warrant further investigation in BUILD-3, a long-term morbidity/mortality phase III study of bosentan in IPF (fig. 9⇓).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The BUILD programme: the future. IPF: idiopathic pulmonary fibrosis; ATS: American Thoracic Society; ERS: European Respiratory Society; CT: computed tomography; 6MWT: 6-min walk test; QoL: quality of life; PFTs: pulmonary function tests.
CONCLUSIONS
Idiopathic pulmonary fibrosis is a severe progressive lung disease, which in the majority of cases is fatal. Although the aetiology remains to be elucidated, the cardinal event in pathogenesis is currently believed to be an aberrant host response to wound healing following alveolar epithelial cell injury. Endothelin, a well-established pathogenic mediator in pulmonary arterial hypertension, is also thought to play a profibrotic role in the pathogenesis of idiopathic pulmonary fibrosis and also idiopathic pulmonary fibrosis-related pulmonary hypertension, a notion that is supported by in vitro experiments. The potential efficacy of the dual endothelin receptor antagonist bosentan is supported by pre-clinical data and bosentan will continue to be evaluated in the treatment of idiopathic pulmonary fibrosis as part of the BUILD clinical trial programme.
Statement of interest
R.M. du Bois has received: reimbursement for attending a symposium; fees for speaking; and fees for consulting. R.M. du Bois has served as a co-chair, advisory board member or consultant to a number of pharmaceutical companies involved in the development of trialling of novel therapies for interstitial lung disease, including Actelion, and has been paid for lecturing at a number of international meetings organised by the pharmaceutical industry, including Actelion.
- © ERSJ Ltd
References









