Article Text

Abstract
Neurofibromatosis has been known to involve blood vessels throughout the body. Pulmonary involvement with interstitial fibrosing alveolitis has been described but no case of pulmonary vascular involvement has been reported to date. A 51 year old patient with cutaneous neurofibromatosis is described who presented with severe pulmonary hypertension and radiographic, scintigraphic, and angiographic evidence of chronic thromboembolic pulmonary hypertension. Severe intimal fibrosis consistent with vascular involvement with neurofibromatosis was found on endarterectomy with no evidence of pulmonary thromboembolism. Neurofibromatosis of pulmonary arteries should be considered as a possible cause of pulmonary hypertension.
- neurofibromatosis
- pulmonary thromboembolism
- intimal fibrosis
Statistics from Altmetric.com
Neurofibromatosis, a common single gene disorder of the nervous system, has been found to involve the lungs with either parenchymal tumours or fibrosing alveolitis.1 We present the case history of a patient with neurofibromatosis who developed severe pulmonary hypertension. Thromboembolic disease was initially suspected because of the findings on lung perfusion-ventilation scanning, spiral CT scanning, and pulmonary arteriography.
Case report
A 51 year old man with neurofibromatosis presented with progressive dyspnoea on exertion (NYHA class 3) whose symptoms progressed during the ensuing year. He had initially been diagnosed elsewhere as having primary pulmonary hypertension and received treatment with warfarin and nifedipine with no improvement in his condition. He denied chest pain, orthopnoea, or paroxysmal nocturnal dyspnoea, or any history of deep vein thrombosis or other thromboembolic disease. He also denied exposure to toxic inhalants, cigarette smoking, or use of appetite suppressants.
Physical examination showed a thin dyspnoeic (24 breaths/min) man without peripheral or central cyanosis. He had numerous cutaneous neurofibromas on both the upper and lower extremities with no cafe au lait pigmentations. No clubbing was found. He had a raised jugular venous pulse and mild pedal oedema. A fixed split second heart sound was heard, while the rest of the physical examination, including the chest, was normal. Laboratory results showed a normal blood count with normal renal and liver function tests. Studies for hypercoagulability were all within normal limits. Arterial blood gas tensions (on room air) were as follows: pH 7.45, Pco 2 32 mm Hg, Po2 57 mm Hg, Sao 291% with an alveolar–arterial gradient of 40 mm Hg.
The chest radiograph showed enlarged pulmonary arteries with no infiltrates or interstitial findings. Electrocardiography showed right ventricular hypertrophy and echocardiography revealed normal left ventricular size and function, dilated right atrium and ventricle, and severe pulmonary hypertension (estimated pulmonary artery gradient of 80–90 mm Hg transoesophageally).
Spirometric tests showed a forced vital capacity (FVC) of 3.39 l (77% predicted), forced expiratory volume in one second (FEV1) of 2.59 l (73% predicted), and FEV1/FVC of 76%. Corrected carbon monoxide transfer factor (Tlco) was 49% of the predicted value. A Doppler ultrasound investigation of the deep leg veins showed no evidence of thrombosis.
A ventilation-perfusion lung scan was performed and revealed large bilateral perfusion defects interpreted as highly probable for diffuse multiple pulmonary emboli. A subsequent spiral CT scan showed multiple bilateral filling defects in the pulmonary arterial tree. Pulmonary angiography was performed, showing multiple peripheral filling defects on the left and central defects in the right main pulmonary artery. Towards the end of this procedure an inferior vena caval filter (bird’s nest) was inserted.
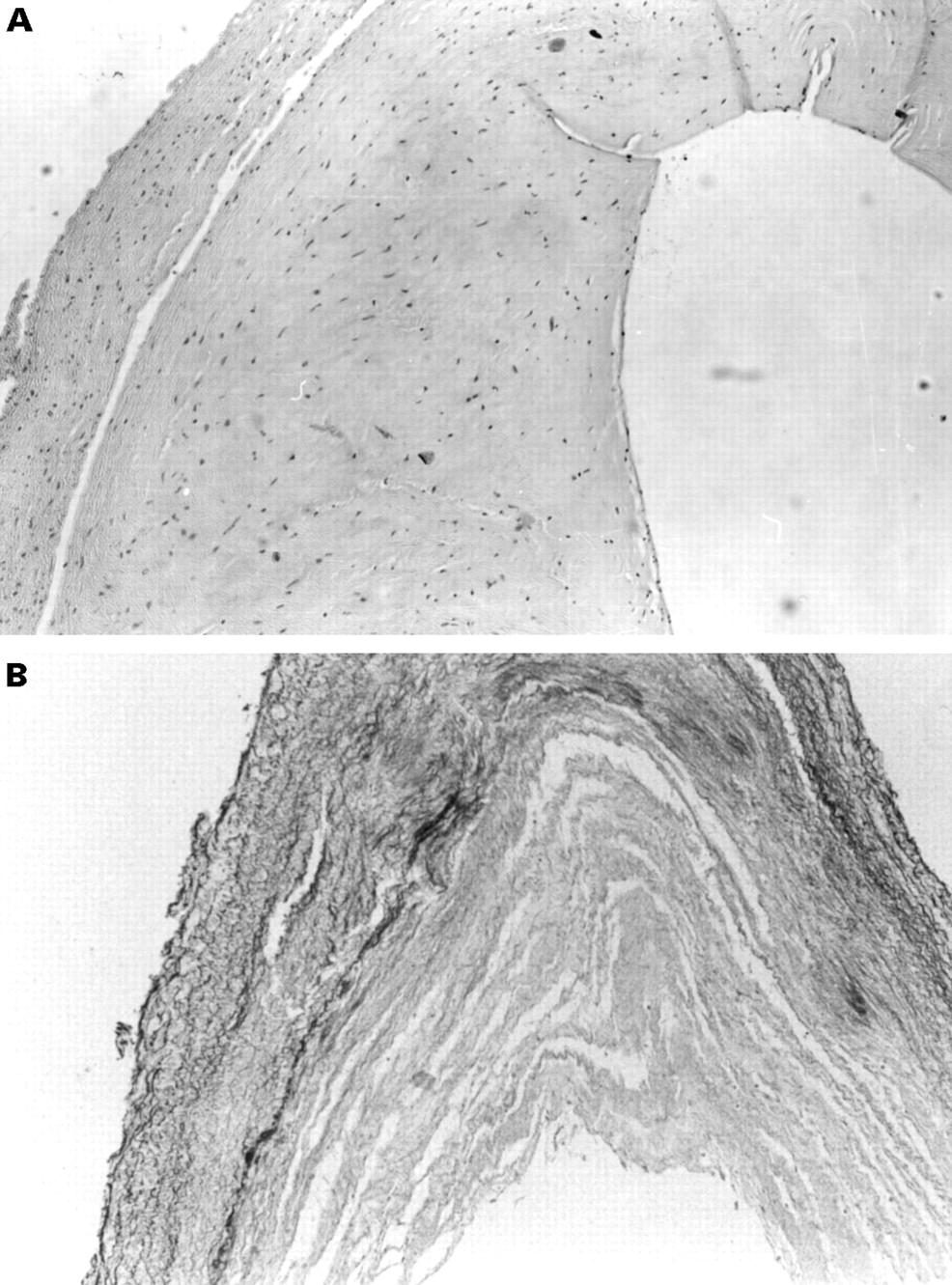
With a diagnosis of suspected major vessel chronic thromboembolic pulmonary hypertension the patient underwent pulmonary thrombendarterectomy of both left and right pulmonary arteries and their major branches. Preoperative catheterisation showed a cardiac index of 3.38 l/min/m2 and a pulmonary vascular resistance index of 236.7 dyne.s.min2/cm5. No evidence of thrombi was found at surgery, while severe diffuse intimal thickening was noted throughout the pulmonary vessels. Because of the widespread involvement of the pulmonary vasculature, stenting of the arteries was not attempted. The pathological specimens taken from the endarterectomy showed extensive irregular thickening of the intima by fibrous tissue (fig 1).
{kind=link}
Microscopic section of a biopsied pulmonary artery. (A) Haematoxylin-eosin stain (× 100) shows a thin rim of media and extensive irregular thickening of the intima by fibrous tissue. No evidence of thromboembolic disease was found. (B) Elastic van Gieson stain (× 100) shows fragmentation of the elastic layers in the media and marked fibrous intimal thickening in another area of the same artery.
The patient was discharged after a protracted postoperative stay in hospital and an echocardiogram performed four months later showed a systolic gradient on the tricuspid valve with an estimated pulmonary arterial pressure of 47 mm Hg. At this time his functional status had also improved (NYHA class 1–2).
Discussion
Pulmonary involvement in patients with neurofibromatosis has been reported with both neurofibromatous tumours within the lung parenchyma and with fibrosing alveolitis.1 In our patient no evidence of either of these conditions was found on CT scanning or by measurement of the Tlco. To the best of our knowledge, involvement of the pulmonary arteries with neurofibromas has not previously been reported although vascular complications of neurofibromatosis have been seen in other organ systems. The retinal,2 lumbar,3 mesenteric,4and renal arteries5 have all been reported to be involved with neurofibromatosis, as have both the intracranial and extracranial arteries.6 The generalised mesenchymal dysplasia of neurofibromatosis predisposes these arteries to rupture or aneurysm formation.6 Biopsy specimens of the arterial wall in these patients have shown marked intimal thickening with proliferation of fibroblastic spindle cells.4 ,5 Histopathological studies have found expression of neurofibromatosis mRNA in cells of the tunica intima of involved blood vessels.7 In addition, rapidly growing neurofibromas show a higher proportion of insulin-like growth factor 1 immunoreactivity in vascular cells (endothelial cells, smooth muscle cells, and fibroblasts) than in slower growing tumours.8
Our patient presented with severe pulmonary hypertension and findings mimicking chronic thromboembolic pulmonary hypertension, a diagnosis supported by the ventilation-perfusion scan, spiral CT scan, and angiography. Biopsy specimens of the arterial wall showed changes consistent with those found in other patients with vascular involvement with neurofibromatosis, changes which apparently caused the vasculo-occlusive picture found in the tests described.
In conclusion, we have described a case of pulmonary hypertension secondary to involvement of the pulmonary arterial tree with intimal fibrosis due to neurofibromatosis. This entity must be considered in the differential diagnosis of pulmonary hypertension found in patients with neurofibromatosis as it can mimic clinical and angiographic evidence of thromboembolic disease. Although endarterectomy resulted in partial improvement in the pulmonary hypertension in our patient, the value of this procedure remains unclear.