Article Text

Abstract
Smoking-related interstitial fibrosis (SRIF) is a common, histologically striking finding in smokers that must be distinguished from the idiopathic interstitial pneumonias and other chronic interstitial fibrosing lesions. It is characterised by marked thickening of alveolar septa by fibrosis composed of thick collagen bundles that have a distinctive hyalinised quality and often are admixed with variable numbers of hyperplastic smooth muscle fibres. There is minimal accompanying inflammation. This fibrosis is usually most prominent in subpleural and centrilobular parenchyma, but can be present elsewhere as well. It is accompanied by emphysema and respiratory bronchiolitis. Most patients are asymptomatic or only mildly symptomatic, and the clinical course is stable in most. This paper reviews the pathologic features of SRIF in detail, its differentiation from more ominous interstitial fibrosing processes, and the clinical implications of its diagnosis.
- fibrosis
- Lung
- Smoking
- Pulmonary Pathology
Statistics from Altmetric.com
Introduction
The chronic interstitial lung diseases consist of a group of clinically and radiographically similar conditions whose diagnosis usually depends on the findings at lung biopsy.1 ,2 Most are idiopathic, and even when an aetiology is identified, as in chronic hypersensitivity pneumonia or collagen vascular disease associated cases, for example, the diagnosis is often first suggested by the pathologist. Classification has expanded in recent years, and can be confusing for pathologists and clinicians alike, yet correct diagnosis is essential, especially to distinguish usual interstitial pneumonia (UIP) and other bad prognosis variants from less lethal conditions.3 ,4 Although addition of yet another variant into the mix may risk further muddling of an already complex classification scheme, smoking-related interstitial fibrosis (SRIF) is a relatively common finding that has distinct histologic features, and it needs to be distinguished from the other fibrosing interstitial lung diseases. This review will describe the pathologic features of SRIF, its differentiation from other fibrosing processes, and its clinical relevance.
SRIF—background
The term, SRIF, was first used in the current context in a report of changes observed in non-neoplastic lung parenchyma in lobectomy specimens from smokers with cancer.5 The authors were struck by the frequent finding of prominent, severe fibrosis with accompanying respiratory bronchiolitis (RB) and emphysema that was apparently unassociated with clinical symptoms. In that study, microscopic slides from 27 sections of non-neoplastic lung parenchyma were examined from each case, thus extensively sampling multiple areas of lung other than tumour. SRIF was found in nine of 20 lobectomy specimens (45%). The lesion was not a minor or focal finding, since for inclusion in the report it had to be present in at least eight of 27 (30%) slides. In fact, it was very extensive in six of nine cases in which it involved 20 or more (74%) slides. One additional similar case was not included because the fibrosis involved only three slides.
SRIF is not a new entity. A similar lesion was reported previously by Yousem6 as ‘respiratory bronchiolitis interstitial lung disease (RBILD) with fibrosis’ in biopsy specimens from nine patients. He additionally identified this lesion in four of 30 (14%) lobectomy specimens from smokers with cancer, while it was absent in 23 pneumonectomy and lobectomy specimens from non-smokers. SRIF also seems similar to ‘airspace enlargement with fibrosis’ described by Kawabata et al7 in 99 of 572 (17.3%) lobectomies from smokers with cancer, and in only one of 230 apparent non-smokers. The more frequent identification of SRIF in Katzenstein et al's5 study compared with both Yousem's6 and Kawabata et al's7 studies may be due, in part, to the extensive sampling that was performed. Another possible explanation is the fact that almost all the lobectomy specimens in the study by Katzenstein et al5 were from the upper (16/20) or middle lobe (2/20) with only two from the lower lobe. SRIF was found only in the upper (8/9) and middle (1/9) lobes, and, therefore, the relatively few lower lobes examined may have skewed the results.
Pathologic features of SRIF
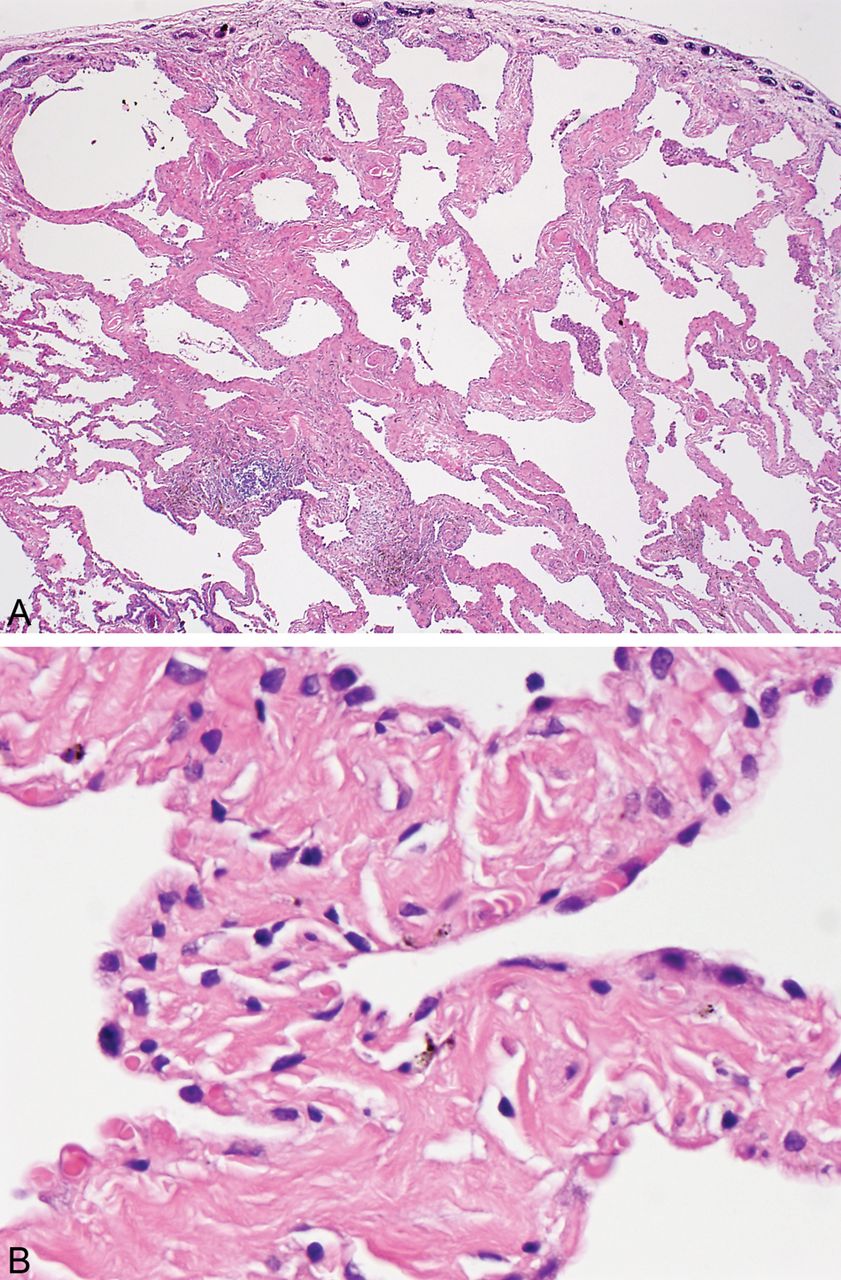
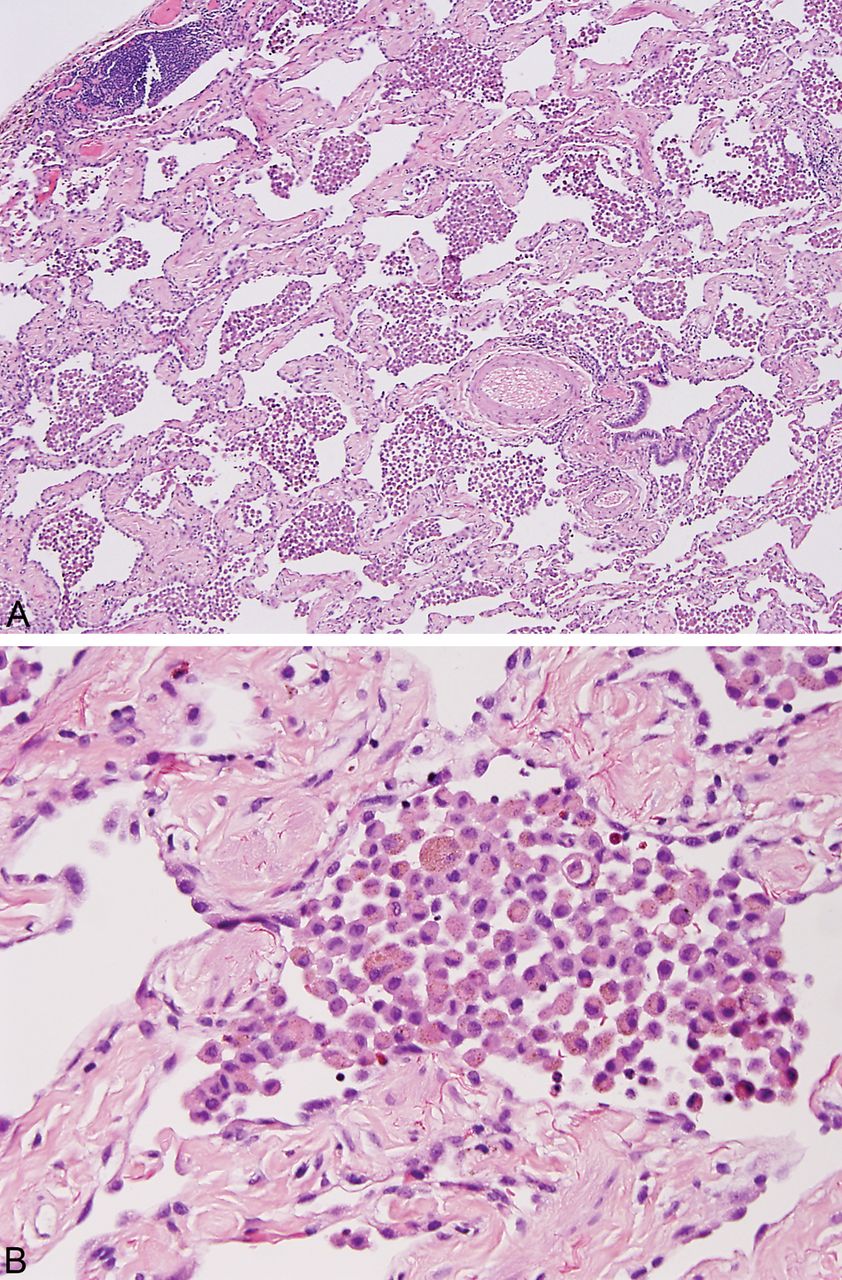
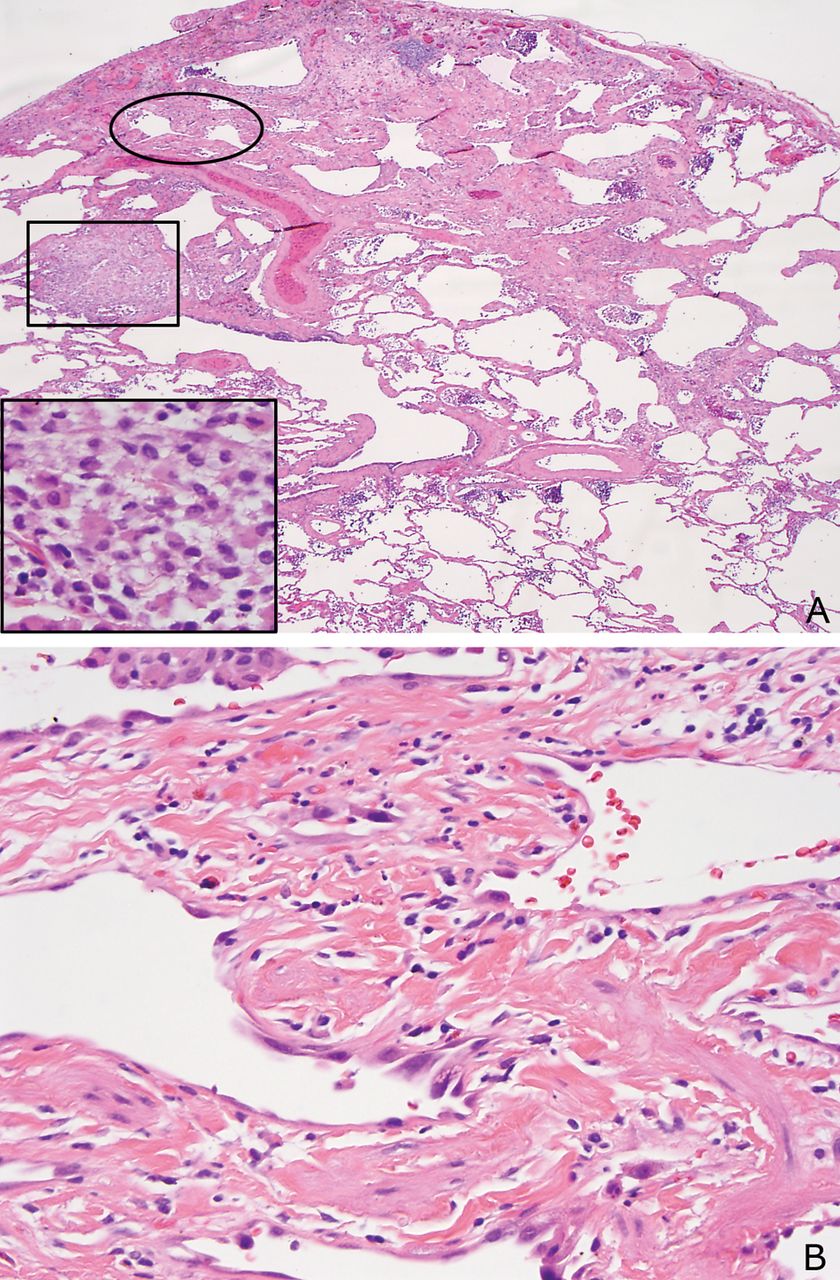
SRIF is characterised by a distinct type of hyalinised interstitial fibrosis that is associated with emphysema and RB.5 ,8 The affected alveolar septa are thickened by deposition of dense, eosinophilic ropey-appearing collagen often with admixed hyperplastic smooth muscle bundles (figure 1). This alveolar septal thickening is usually quite marked and easily visible at low magnification. It tends to be most prominent in subpleural parenchyma where it is associated with emphysema, but it can also be found in deeper parenchyma including both centrilobular and other more random areas unassociated with emphysema (figure 2). The appearance contrasts sharply with the mild fibrosis that commonly accompanies examples of ordinary emphysema (figure 3). Intra-alveolar pigmented macrophages (smokers’ macrophages) that are indicative of RB are invariably present and usually numerous. They may be so numerous in some cases as to fill the alveolar spaces adjacent to the thickened alveolar septa (figure 4). Occasional foci of mild chronic inflammation are admixed with the interstitial collagen, but inflammation is never prominent or diffusely present, and in most areas the collagen is devoid of inflammation.

(A) Low magnification view of smoking-related interstitial fibrosis (SRIF) showing the characteristic marked thickening of alveolar septa in subpleural parenchyma associated with emphysema. Clusters of pigmented macrophages indicative of RB are present in some airspaces. The pleural surface is on the top. (B) High magnification view of same case showing the thick, ropey, hyalinised collagen deposition within alveolar septa typical of SRIF.

(A) Low magnification view of smoking-related interstitial fibrosis within deeper lung parenchyma. (B) Higher magnification showing typical eosinophilic collagen deposition along with entrapped, hyperplastic smooth muscle bundles.

(A) Low magnification view of typical centrilobular emphysema showing mild fibrosis in the wall of the emphysematous space. This type of fibrosis is common in ordinary emphysema and does not represent smoking-related interstitial fibrosis (SRIF). (B) In this field from the same case and at the same magnification there is prominent alveolar septal collagen deposition associated with mild emphysema that is characteristic of SRIF. Numerous pigmented macrophages (RB) are also present in airspaces.

In this example of smoking-related interstitial fibrosis the airspaces are filled with pigmented macrophages (RB), a finding that is reminiscent of desquamative interstitial pneumonia (DIP). (A) Low magnification with pleural surface at upper left. (B) High magnification illustrating the hyalinised collagen and smooth muscle bundles within alveolar septa and the pigmented intra-alveolar macrophages. This marked interstitial fibrosis along with the associated emphysema excludes DIP.
Differential diagnosis
Usual interstitial pneumonia
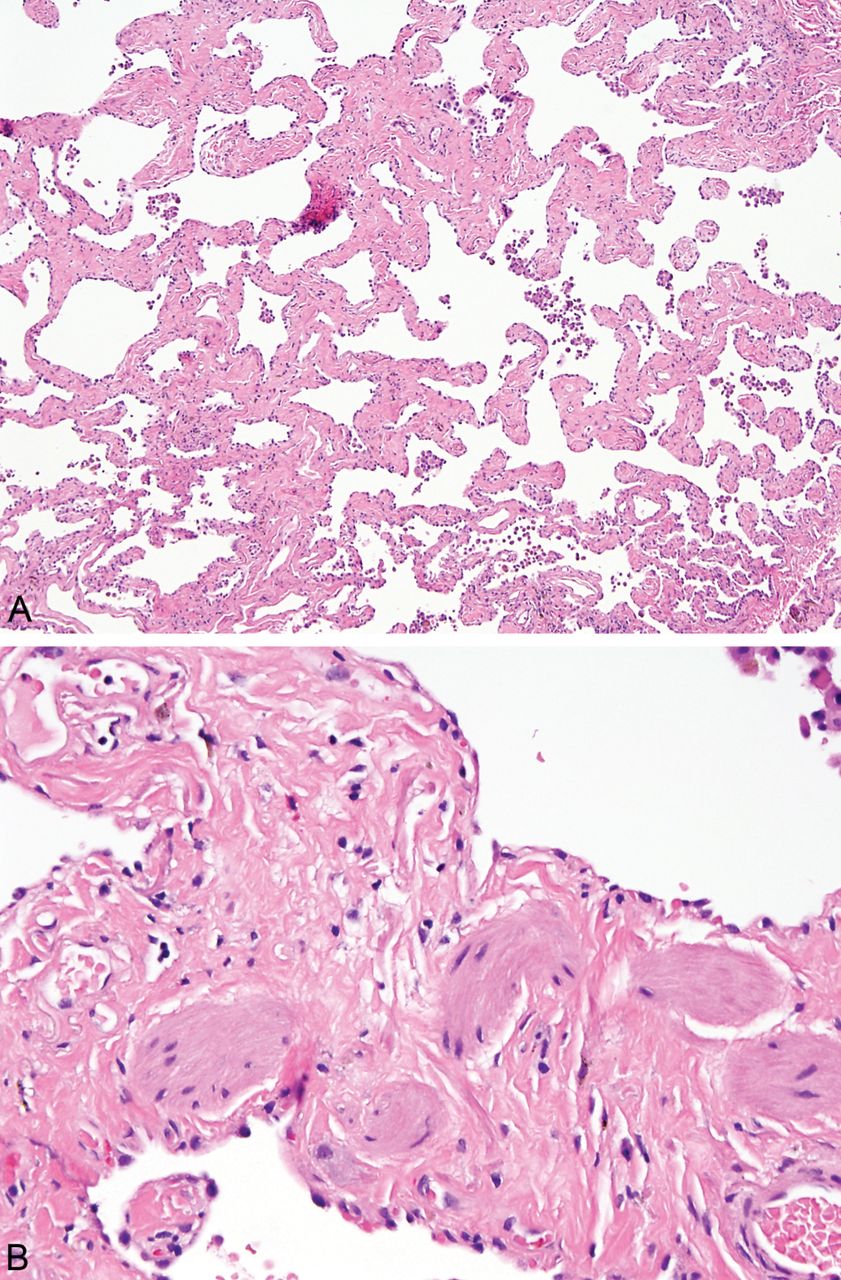
SRIF differs from UIP in several ways, and the distinction is usually not difficult.3 ,8 First, well-formed honeycomb change, defined as enlarged airspaces lined by bronchiolar type epithelium and often containing mucin and inflammatory cells, which is present in most cases of UIP, is not a feature of SRIF (figure 5). Second, the hyalinised fibrosis so characteristic of SRIF is not a characteristic feature of UIP. Third, although occasional fibroblast foci may be found in SRIF, they are never prominent. Finally, the constant association of SRIF with emphysema and RB further aids in the differential diagnosis.

Photomicrographs contrasting the appearance of the honey-comb change of usual interstitial pneumonia (UIP) with the fibrosis and architectural distortion of smoking-related interstitial fibrosis (SRIF). (A) Low magnification of honeycomb change in UIP showing enlarged airspaces filled with a mucinous exudate and partially lined by bronchiolar epithelium. The underlying lung architecture is destroyed by scarring and chronic inflammation. Inset is a higher magnification showing the bronchiolar epithelium lining the honeycomb space. (B) Low magnification view of SRIF showing the typical enlarged (emphysematous) airspaces lined by densely fibrotic alveolar septa. Note the empty spaces and lack of bronchiolar type epithelial lining. Inset is a high magnification of the typical collagen deposition with minimal inflammation. It is easy to see how this kind of subpleural airspace enlargement with fibrosis may cause radiographic findings mimicking honeycomb change.
Fibrosing non-specific interstitial pneumonia
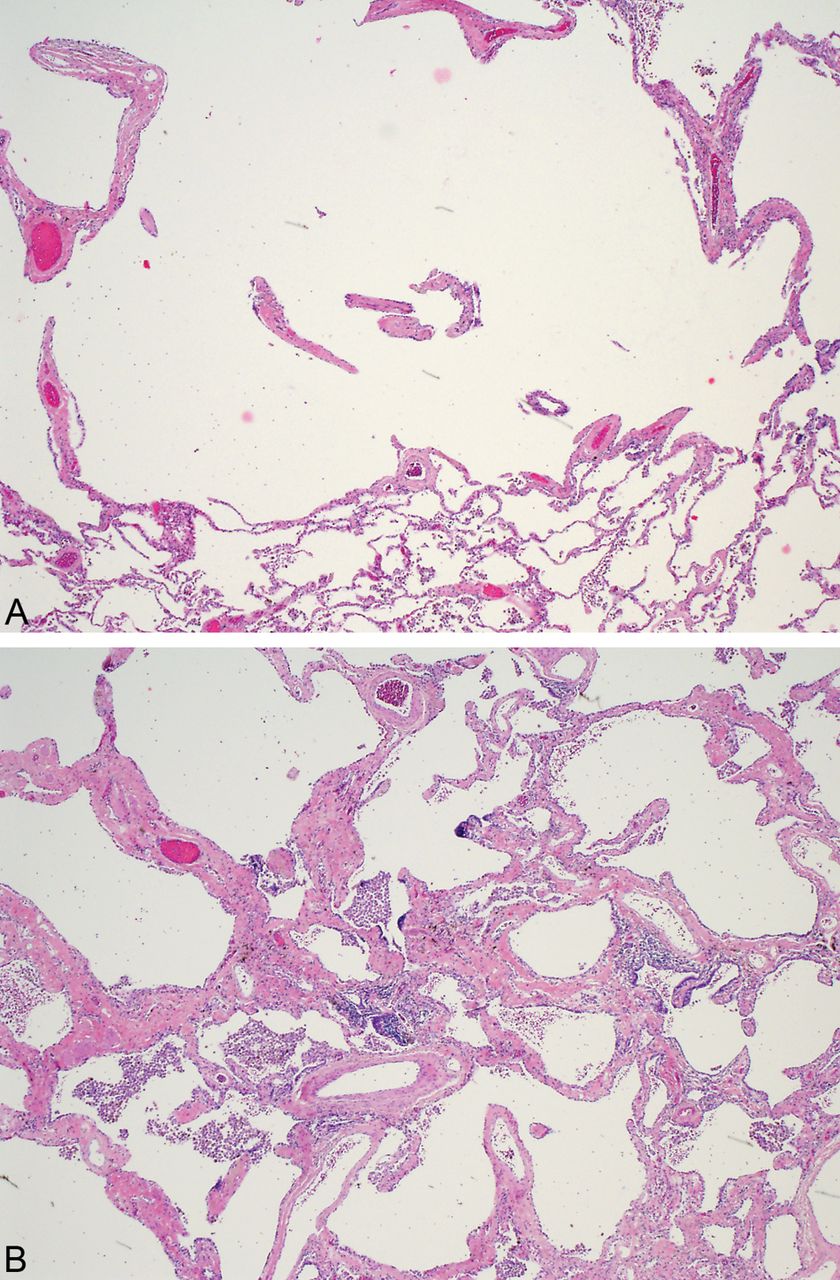
The distinction of fibrosing non-specific interstitial pneumonia (NSIP) from SRIF can be challenging, but attention to the appearance of the fibrosis, the amount of accompanying inflammation and the distribution of the changes should lead to accurate diagnosis.1 ,9 The deeply eosinophilic, hyalinised collagen of SRIF contrasts with the loose collagen fibrosis typical of NSIP and most other chronic fibrosing disorders (figure 6). Even when NSIP becomes severely fibrotic, chronic inflammatory cells are usually present to some degree admixed with the collagen, whereas in SRIF there are areas devoid of inflammatory cells. Fibrosing NSIP usually has a fairly diffuse distribution which contrasts with the preferential subpleural and centrilobular accentuation characteristic of SRIF. Emphysema is usually not a prominent feature in NSIP, although RB may be present if the patients are smokers.

Photomicrograph contrasting the interstitial fibrosis of fibrosing NSIP with smoking-related interstitial fibrosis (SRIF). (A) NSIP, showing loose alveolar septal fibrosis with intermingled chronic inflammatory cells. (B) SRIF with dense, eosinophilic collagen and minimal inflammation.
RBILD/Desquamative Interstitial Pneumonia (DIP)
RBILD and DIP are considered by many to represent different ends of a spectrum of the same disease that occurs in smokers and is characterised by increased numbers of pigmented (smokers’) macrophages within airspaces.1 ,10 ‘DIP’ is an outmoded name that is not conceptually correct (the intra-alveolar cells are not desquamated pneumocytes), and since there are no firm distinguishing features from RBILD, we prefer to use the term RBILD for the entire spectrum. The diagnosis of RBILD (and ‘DIP’) has evolved in recent years to include only those cases with intra-alveolar smokers’ macrophages and only minimal, if any, interstitial fibrosis. The distinction from SRIF, therefore, is not difficult, since the striking fibrosis of SRIF excludes RBILD (figure 4).
Scarred Langerhans Cell Histiocytosis (LCH)
Since patients with LCH are smokers, it is common to find other smoking-related changes in biopsies showing LCH, including RB, emphysema and SRIF. The diagnosis of LCH requires nodular interstitial infiltrates of Langerhans cells, but as the disease progresses the cellular nodules can be replaced by dense, often hyalinised fibrosis.1 The distinguishing feature of these scarred areas is that they usually are peribronchiolar and stellate shaped, and they often are surrounded by traction emphysema, all features that differ from SRIF. In some cases, however, they do blend into a more uniform thickening of alveolar septa with hyalinised fibrosis typical of SRIF (figure 7). Whether SRIF and scarred LCH in such cases are independent manifestations of smoking that happen to occur in the same patient, or whether SRIF is somehow related to the scarring of LCH is uncertain.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Photomicrograph showing smoking-related interstitial fibrosis (SRIF) in a case of Langerhans cell histiocytosis (LCH). (A) Low magnification view showing typical SRIF in subpleural parenchyma and a nearby nodule of LCH. Inset is a high magnification of the LCH nodule (rectangle) showing the diagnostic Langerhans cell infiltrate. (B) High magnification from the circled area of SRIF showing the characteristic collagen deposition with smooth muscle bundles. Scattered neutrophils are present as well, likely related to the surgery.
Combined emphysema and pulmonary fibrosis
Not surprisingly, cases of simultaneous emphysema and pulmonary fibrosis have been described in smokers.11–13 They are defined radiographically by the presence of classic features of idiopathic pulmonary fibrosis (IPF)/UIP in the lower lobes and emphysema in the upper lobes. Although biopsies are usually not performed, most cases likely represent the coincidental occurrence of two fairly common diseases, UIP and emphysema. However, biopsies from a few cases have been reported as showing DIP, ‘variant DIP’, or unclassified interstitial pneumonia, and one illustrated case showed typical features of SRIF.11–13 It seems likely, therefore, that at least some cases designated as combined emphysema and pulmonary fibrosis represent SRIF. The degree to which SRIF cases are included under the rubric of chonic obstructive pulmonary disease (COPD) is another question.
Clinical significance
In most patients, SRIF is an incidental finding in lung tissue removed for other reasons. The average age in Katzenstein et al's5 study was 65 years with a range from 52 years to 77 years. All were either current or ex-smokers, with pack-years smoked ranging from 16 to 80 (mean, 48). There was no gender difference with five men and four women. None had specific symptoms attributable to SRIF, and no changes were identified on chest CT exams that could be attributed to SRIF. However, only one CT was high resolution, and, of course, the exams were focused on the tumours and not the non-neoplastic parenchyma. Mild to moderate obstructive defects were noted in six of eight patients on pulmonary function testing, and there was mild to moderate reduction in diffusion capacity in four. All patients were alive at two to 27 months following surgery (mean follow-up, 16.3 months), and none had evidence of progressive respiratory impairment.
Rarely, patients with SRIF present with respiratory complaints leading to biopsy. The nine such patients reported by Yousem6 were younger than those in Katzenstein et al's5 study, with a mean age of 44 years, and a range from 32 years to 68 years. There were five women and four men with an average pack-years smoked of 35 (range, 15–60). Shortness of breath and/or cough were the most common complaints, and high-resolution CT (HRCT) exams most often showed bilateral micronodular infiltrates and/or ground glass opacities. All patients were alive after a mean follow-up of 3.2 years (range, 0.5–5.2), including seven with stable and two with slowly progressive disease.
Discussion
SRIF is a form of chronic interstitial fibrosis that is clearly definable pathologically and readily distinguished from other types of interstitial fibrosis. The introduction of a new term to an already complex diagnostic armamentarium is justifiable only if the term is relevant, useful and necessary, and SRIF seems to fulfil these requirements. It synthesises several different pathologic changes into a single, readily understandable entity with clear-cut aetiology. Its pathologic findings are straightforward, usually quite striking, and generally easy to distinguish from findings in other fibrosing lung diseases. When the dense fibrosis of SRIF surrounds enlarged airspaces of emphysema in subpleural parenchyma, the changes radiographically can mimic honeycomb change of UIP. It is especially important, therefore, in such cases to recognise SRIF and to distinguish it from UIP and other idiopathic interstitial pneumonias.
Biopsy is only rarely undertaken in patients with SRIF because symptoms are usually mild and overshadowed by other problems such as lung cancer. SRIF is more commonly encountered by pathologists when the non-neoplastic parenchyma is examined from lobectomy specimens removed for cancer. It is important in such cases not to confuse the findings of SRIF with those of UIP or other fibrosing interstitial pneumonias. It seems likely that SRIF will be encountered by pathologists on biopsy specimens more frequently in the future because increasingly sensitive radiographic techniques will identify more and subtler lung abnormalities. Several large screening studies using HRCT already have reported interstitial abnormalities in a significant proportion (2.2–22%) of otherwise asymptomatic smokers.14–16 As new treatment options become available for UIP and other entities, there will be greater impetus for early diagnosis, and it will be increasingly important to sort out SRIF from other causes of radiographically non-classifiable abnormalities, especially UIP.
Although some cases of SRIF have, in the past, been included under the rubric of DIP, SRIF should be separated from that entity because it differs pathologically and also because it is not a type of idiopathic interstitial pneumonia. Furthermore, recognition of SRIF as a specific entity suggests that the time may have come to eliminate DIP from interstitial lung disease terminology altogether. DIP a misnomer, and the term has been applied indiscriminately and incorrectly to several unrelated entities when they occur in smokers, including UIP, NSIP and LCH, for example. Intra-alveolar macrophage accumulation, although often a striking finding in those conditions as in SRIF, simply reflects the fact that the patient is a cigarette smoker, and it is unrelated to the underlying disease.
Since emphysema accompanies the fibrosis of SRIF, a logical question is whether SRIF is simply a ‘normal’ finding in severe emphysema. Although most classic descriptions of emphysema do not mention or illustrate fibrosis,17 ,18 it is widely accepted that some fibrosis commonly accompanies emphysema.19–22 In fact, emphysema is defined as ‘abnormal, permanent enlargement of airspaces distal to the terminal bronchioles, accompanied by destruction of their walls and without obvious gross fibrosis’.23 The ‘without obvious gross fibrosis’ was added to earlier definitions to reflect the fact that some fibrosis may be present microscopically, although not to the degree that would be visible grossly. The extent of the fibrosis in SRIF with the associated marked alveolar wall thickening is clearly greater than the mild fibrosis that commonly accompanies emphysema. The fibrosis of SRIF further differs from the mild fibrosis of emphysema in that it can occur in non-emphysematous parenchyma, and it can be identified grossly. Therefore, although SRIF likely is related in some way to emphysema, it is not a typical feature, and it needs to be recognised by pathologists so that it is not misdiagnosed as a fibrosing interstitial pneumonia.
Take home messages
-
Smoking-related interstitial fibrosis (SRIF) is a distinct form of interstitial fibrosis with a striking pathologic appearance that occurs in smokers and is characterised by hyalinised fibrosis thickening alveolar septa associated with respiratory bronchiolitis and emphysema.
-
Most cases of SRIF are incidental, and biopsies are undertaken in only a few cases. The lesion is likely to be encountered with increasing frequency, however, as sophisticated radiographic screening procedures become more widely used.
-
SRIF needs to be distinguished from usual interstitial pneumonia and other idiopathic interstitial pneumonias which it can mimic clinically and radiographically.
-
SRIF may be the underlying cause of the fibrosis in some cases of combined fibrosis and emphysema. Its role in chronic obstructive pulmonary disease (COPD) is unknown.
References
Footnotes
-
Competing interests None.
-
Provenance and peer review Commissioned; internally peer reviewed.