Article Text

Abstract
Primary pulmonary hypertension (PPH) represents the end stage of a disruption of pulmonary vascular integrity, of unknown cause. Although PPH is associated with several systemic disorders, there have hitherto been few clues as to the aetiological factors responsible for the pathogenesis of this condition. As an example of the application of modern molecular genetics and positional cloning, this leader desribes the range of studies currently under way, which aim to find the gene that underlies PPH, and summarises the implications of the identification of such a gene.
- pulmonary hypertension
- genetics
- chromosome 2q33
Statistics from Altmetric.com
Pulmonary arterial hypertension is a devastating disease, characterised clinically by a raised pulmonary artery pressure (mean > 25 mm Hg at rest or > 30 mm Hg during exercise), with normal pulmonary wedge pressure, and subsequent right heart failure.1 Pulmonary arterial hypertension has been classified as: (1) primary pulmonary hypertension (PPH), of unknown cause, which is either sporadic or familial (at least two family members); and (2) related to other conditions such as human immunodeficiency virus (HIV) infection and ingestion of appetite suppressants. There are many secondary causes of raised pulmonary artery pressure, including chronic obstructive pulmonary disease and thromboemboli, which are listed fully in table 1, in accord with the 1998 World Health Organisation classification of pulmonary hypertension.2 This review will focus on our present understanding of the clinical and molecular genetics of PPH and its relation to other forms of pulmonary arterial hypertension.
WHO classification of pulmonary hypertension
PPH has a reported incidence of one to two new diagnoses/million/year in developed countries and is twice as common in women as in men, although it has a similar disease progression in both sexes. The mean age of onset is 36.4 years but it can occur at any stage of life and it has a median survival time of 2.8–3.4 years from diagnosis.3–5 Diagnosis of the disease can be lengthy, in part because of the non-specific presentation with breathlessness and fatigue, few clinical signs, and the numerous tests required to exclude secondary causes of pulmonary hypertension.1,3 Treatment is based on calcium antagonists and anticoagulants, with patients progressing to prostacyclin analogues and some to heart–lung transplantation.6,7J Clin Pathol 2000;53:899–903
Clinical genetics of familial PPH
Dresdale first described PPH in 1951, and its heritability in 1954.8,9 Although most cases are apparently “sporadic”, one series showed that 6% of patients have at least one other family member with the condition.3 We have now identified 21 affected families throughout the UK. Over 100 families have been recognised in the USA with further kindreds described in Europe, Japan, and Australia.
Clinical studies of families with PPH have revealed several important features. Many examples of male to male transmission of PPH have been cited, excluding X linked inheritance, and vertical transmission supports autosomal dominant segregation (MIM 178600).10,11 Many PPH kindreds show generation “skips” in the expression of the disease, reflecting reduced “penetrance” of the gene, which is estimated to be as low as 20% (that is, 80% of those who inherit the abnormal gene will not develop symptoms of the disease) (fig 1). Previous descriptions of two or more siblings with PPH have suggested recessive inheritance (MIM 265400).11–13 However, this is more likely to represent the reduced gene penetrance, pointing to a role and requirement for additional factors, both genetic and environmental, in the generation of symptomatic PPH.

Familial primary pulmonary hypertension (PPH). A hypothetical pedigree illustrating reduced genetic penetrance of the disease (that is, skipping of generations) as seen in individuals II:2 and III:2, “anticipation” (earlier age of onset in successive generations), and the female preponderance. Open square, unaffected male; open circle, unaffected female; closed square, affected male; closed circle, affected female; slash, deceased.
Longer term follow up of families with PPH has provoked debate about the decreasing age of onset of disease in subsequent generations; a feature referred to as “anticipation” (fig 1).14 One biological explanation of this phenomenon is the unstable expansion of DNA trinucleotide repeat sequences. Approximately 10% of the human genome is composed of repetitive DNA repeats, which are usually inherited in a stable form. Certain trinucleotide repeats become unstable and prone to dramatic expansion, which affects the expression of neighbouring genes. The instability of these repeats means that different members of the same family can show different repeat lengths and corresponding variation in clinical severity.
Interestingly, to date, trinucleotide repeat expansion has been implicated only in conditions with a neurological component—for example Huntingdon's chorea and myotonic dystrophy—and could lead to abnormal gene function in PPH through erroneous neuronal control of pulmonary vascular resistance.15 However, these observations in PPH kindreds are more likely to have arisen from pronounced ascertainment bias through the impact of screening programmes, rather than being the consequence of genetically determined variation. An alternative hypothesis is that “at risk” individuals are receiving earlier and heavier exposure to an environmental agent responsible for the promotion of disease.
Molecular genetics of familial PPH
Striking success has been achieved in the identification of the genes contributing to inherited cardiomyopathies through a “candidate gene” approach.16 This relies upon knowledge of specific proteins involved in the underlying pathology of the diseases. PPH genetics has necessitated a different strategy for gene identification, that of disease gene “positional cloning”. In 1997, two groups working independently, Nichols et al and Morse et al, performed a genome wide search for the PPH gene using DNA markers in a total of seven families with PPH. Linkage was established on the long arm of chromosome 2 and the gene was placed between two DNA markers, D2S335 and D2S369, 25 000 000 bp apart.17,18 To date, all affected families studied are linked to this gene locus, termed PPH1, implying one predisposing disease gene only. Knowledge of the disease haplotype has been used by Morse et al to detect at risk individuals in one family with PPH.19 The use of genetic markers in predictive testing depends on the availability of samples from family members sufficient to demonstrate linkage, in addition to homogeneity for the disease locus, and we would caution the use of this approach pending gene identification.
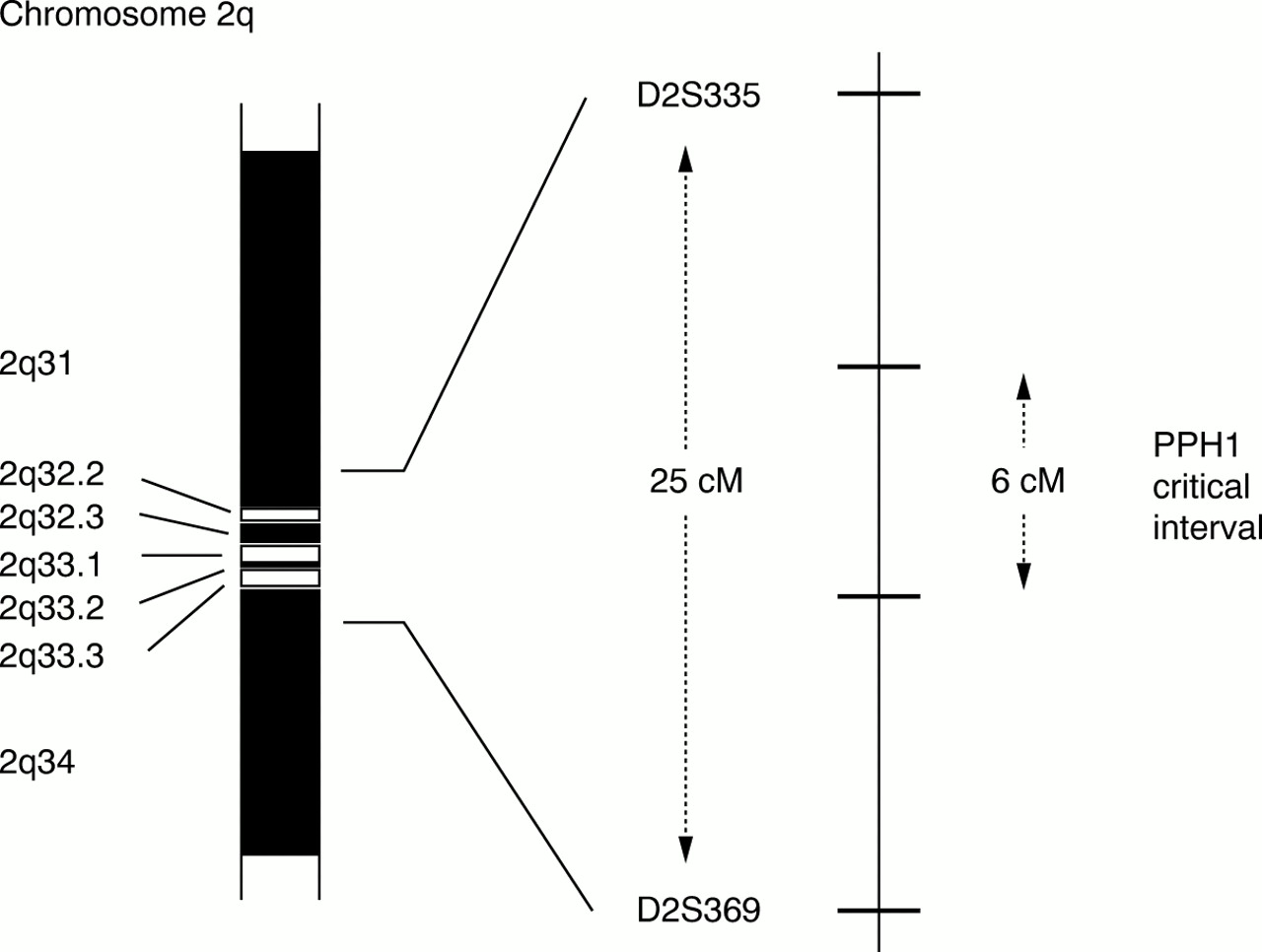
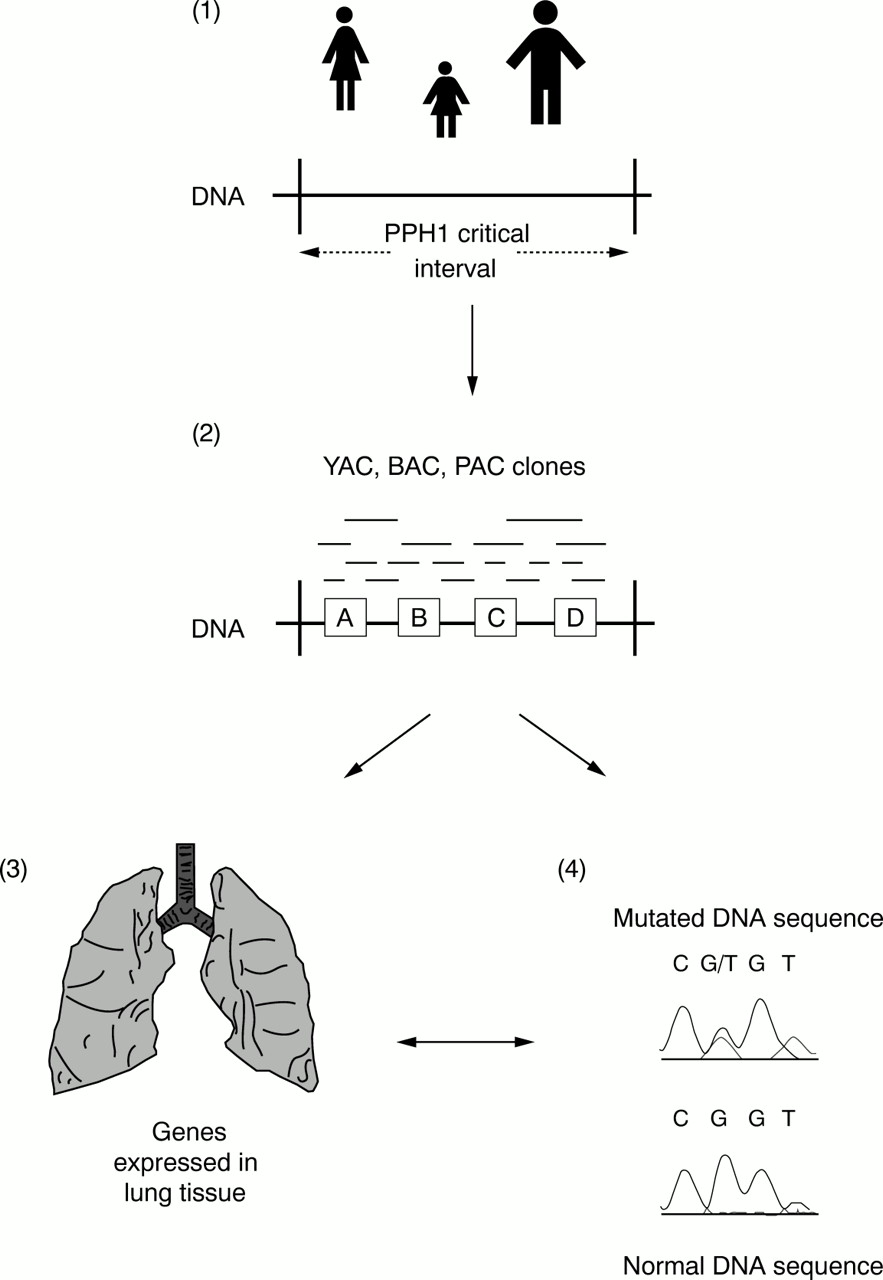
The PPH1 genetic interval has been reduced from the original 25 000 000 bp of DNA, through the recognition of recombination events in PPH families, but still spans at least 6000 000 bp containing over 50 putative genes (fig 2).20 We have placed 17 previously identified genes within this interval, each representing a positional candidate for the PPH gene.20a Of these, several are plausible candidates, including usurpin and caspases 8 and 10, which are involved in T cell apoptosis and autoimmunity, together with a member of the transforming growth factor β (TGF-β) superfamily. Expression of TGF-β is upregulated in remodelling pulmonary arteries of patients with PPH.21 The identification of the PPH1 gene will require yet greater reduction of the genetic critical interval, facilitated by the identification of further families with PPH and the establishment of the International PPH Consortium*. The strategy used to characterise the PPH gene is outlined in fig 3. Re-evaluation of the pathobiology of PPH might help guide investigators as to the nature of the gene they seek.

Idiogram of the long arm of chromosome 2 (2q). The PPH1 gene location showing genetic markers D2S335 and D2S369 and the reduction of the critical interval from 25 to 6 centimorgans (cM).

Idealised positional cloning strategy to identify the PPH1 gene. (1) First, further families with primary pulmonary hypertension (PPH) are required to reduce the PPH1 critical interval by the detection of recombinant events; that is, identifying that part of 2q33 shared by affected individuals within a family. (2) Use of data from the human genome project to identify genes and expressed sequence tags (ESTs or partially sequenced genes) that appear to be contained within the PPH1 critical interval (for example, genes A, B, C, and D). To place these genes, ESTs, and other DNA markers in the correct order, a series of overlapping DNA clones inserted into yeast, bacteria, or plasmid artificial chromosomes (YACs, BACs, and PACs, respectively) is created: a so called physical map. (3) Studies are undertaken to identify which of these genes are expressed in lung tissue and hence are good candidates for PPH1. (4) Finally, direct analysis of genes to detect a mutation in affected individuals compared with normal controls.
Pathobiology of PPH providing genetic clues
PPH is associated with narrowing of the precapillary pulmonary arterioles, reflecting endothelial and smooth muscle cell proliferation, pronounced vasoconstriction, and in situ thrombus formation. Abnormal expression of ion channels, altered concentrations of vasoactive mediators, aberrant vascular remodelling in response to haemodynamic change, and pulmonary artery endothelial and smooth muscle cell dysfunction may each underlie the molecular mechanisms generating the pathological cascade.22–24 The observed increase in endothelin-1 expression (vasoconstrictor), in combination with reduced urinary excretion of prostacyclin metabolites and endothelial nitric oxide synthase expression in pulmonary arteries of patients with PPH (vasodilators), provides a scientific basis for vasodilator treatment in this disease.25,26
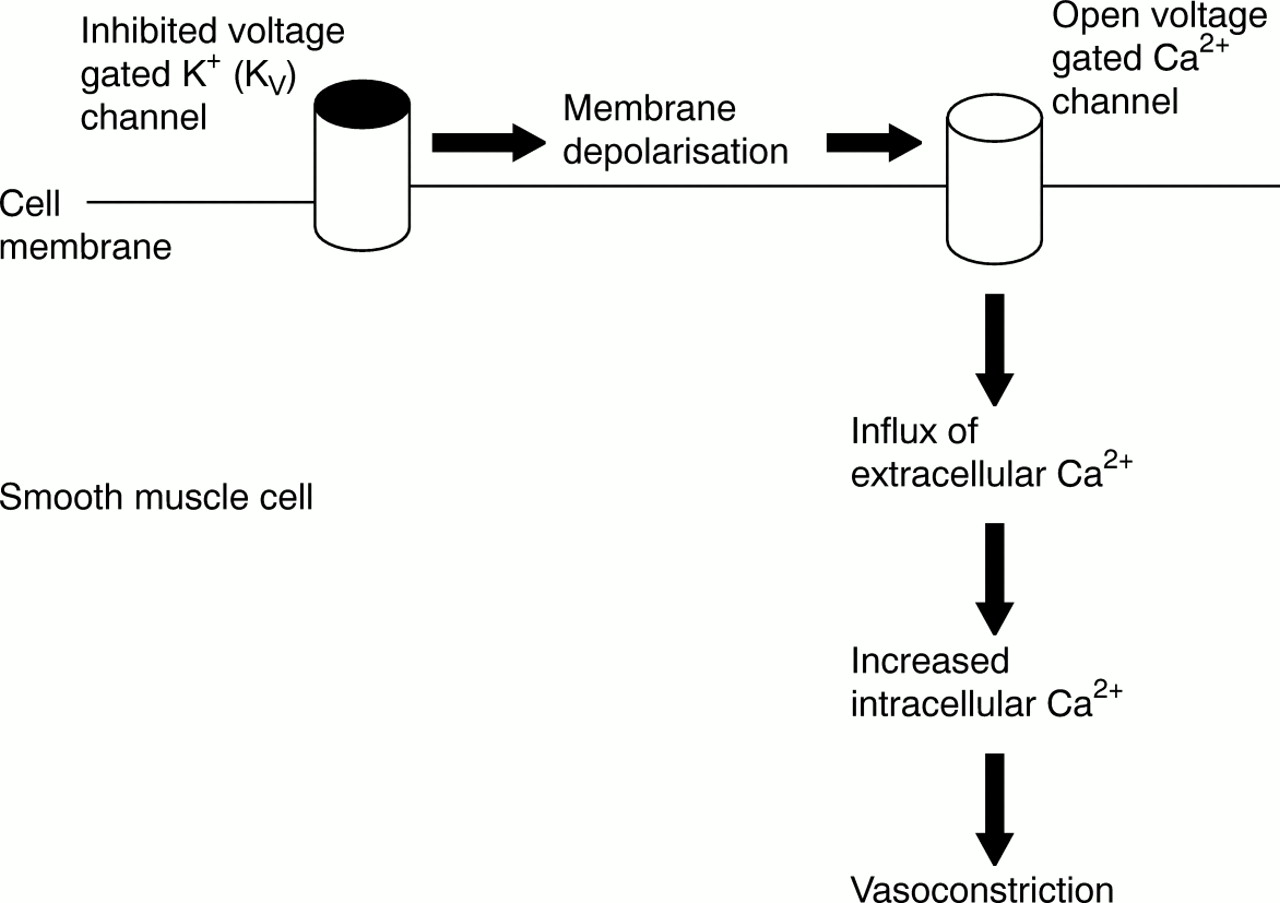
Cytosolic free calcium acts as an important regulator of smooth muscle contraction and cell proliferation. Yuan et al have shown that the opening of voltage gated potassium (KV) channels in pulmonary artery smooth muscle cells of patients with PPH is inhibited, causing membrane depolarisation and a rise in cytosolic calcium, and hence vasoconstriction (fig 4).27 KV channels are composed of pore forming α subunits (for example, KV1.5) and cytoplasmic regulatory subunits (for example, KV1.1). In PPH, KV1.5 mRNA is reduced in comparison with normal and secondary pulmonary hypertension controls.28 Although the gene encoding Kv1.5 is not itself within the PPH1 critical interval, the PPH gene might still be involved with the regulation of these channels.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Inhibition of voltage gated potassium channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension (PPH) causing membrane potential depolarisation, a rise in intracellular calcium, and vasoconstriction.
Further studies of KV channels have exploited the association of ingestion of phenylethylamine based appetite suppressants and the development of pulmonary hypertension. The incidence of pulmonary hypertension in Europe escalated in the 1960s after the release of aminorex fumarate and again in the early 1990s, reflecting the availability of fenfluramine and its d-isomer, dexfenfluramine.29,30 Use of these amphetamine-like drugs for more than three months is associated with a 23.1-fold increased risk of developing pulmonary hypertension, which is clinically and histologically indistinguishable from PPH.31 Wang et al have demonstrated reduced expression of the KV1.5 α subunit in human normotensive pulmonary artery smooth muscle cells after the addition of fenfluramine, supporting a proposal that anorectics might cause pulmonary hypertension through their effect on KV channel gene regulation.32
Further pointers to the genetic basis of PPH have come from clonality studies of the characteristic plexiform lesion. Plexiform lesions are composed of abnormally large endothelial cells seen at pulmonary vessel bifurcations. Voelkel et al examined 22 plexiform lesions from patients with PPH and 20 from patients with secondary pulmonary hypertension.33,34 At the time of reporting, there were strong indications that most plexiform lesions from patients with PPH were monoclonal in origin. Conversely, all plexiform lesions from patients with secondary pulmonary hypertension displayed polyclonal proliferation. These results indicate autonomous growth of endothelial cells in PPH, similar to tumorigenesis and that seen in smooth muscle cells of atherosclerotic plaques, perhaps through abnormal regulation of a gene controlling cell growth.35
This finding has also provoked a “two hit” theory for the molecular development of PPH. In this model, genetic mutations would need to occur on both copies of a PPH gene, as seen with Knudson's two hit hypothesis and the inheritance of retinoblastoma.36 Here, in individuals with familial PPH, one mutation would have to be inherited in the germ line, and exposure to an environmental stimulus would provide the second “somatic” hit in a cell causing monoclonal growth.
Other associated diseases
In 1987, Kim and Factor described the first case of HIV associated pulmonary hypertension.37 By 1997, 88 patients with HIV or AIDS had been described in whom pulmonary hypertension had developed, 42% having previously used intravenous drugs, and with no correlation with CD4 count.38 Pulmonary hypertension is reported to occur in 0.5% of those with HIV infection and follows a similar clinical course with comparable pathological changes to PPH in the absence of HIV.39 These observations support the role of a pulmonary hypertension susceptibility gene. However, despite the rapidly increasing prevalence of HIV infection in developing countries, pulmonary hypertension remains rare, either reflecting a lower frequency of an abnormal gene in these populations or underdiagnosis because of the rapid progression of AIDS and death.
In 1981, a major epidemic of pulmonary hypertension occurred in Spain after the ingestion of adulterated rapeseed oil.40 More than 20 000 people suffered acute pneumonitis, myalgias, and eosinophilia, with 20% developing pulmonary hypertension. The pulmonary hypertension regressed in all but 1.5% of patients up to four years after the initial illness. In those whom pulmonary hypertension progressed, the clinical and histological findings were indistinguishable from PPH. Some of the puzzles surrounding the toxic oil syndrome may be answered through identification of the PPH1 gene, as with HIV infection and appetite suppressants. Those individuals developing persistent pulmonary hypertension subsequent to an environmental agent might only do so if they are genetically susceptible.
Future prospects
Identification of the PPH1 gene will herald a new era of pulmonary vascular biology research. Prospects for the future include the development of more specific and effective treatments, leading to improvement in disease prognosis. In families with PPH, presymptomatic genetic testing will highlight those relatives requiring clinical screening, with the potential benefits of early treatment and avoidance of pregnancy (which may precipitate PPH), environmental “triggers”, and hypoxic environments. In those who have developed pulmonary hypertension following the ingestion of appetite suppressants, toxic oil, and HIV infection, it will become possible to establish whether they have a similar genetic susceptibility, which should prompt further investigation of gene–environment interactions. Through the study of familial PPH a greater understanding of other forms of pulmonary hypertension is likely to follow.
Acknowledgments
The authors gratefully acknowledge the generous support of those with PPH and their families who have contributed to the studies referred to in this article, and funding by the British Heart Foundation (project grant: RCT) and Medical Research Council (MRC clinical training fellowship: JT).
References
Footnotes
↵* The International PPH Consortium: RC Trembath, Division of Medical Genetics, University of Leicester, Leicester LE1 7RH, UK; WC Nichols, Division of Human Genetics, Children's Hospital Medical Center, 3333 Burnet Avenue, Cincinnati OH 45229, USA; T Faroud, Department of Medical and Molecular Genetics, Indiana University School of Medicine, 975 W. Walnut Street IB-155, Indianapolis, IN 46202–5251, USA; JE Loyd, JA Phillips III, JH Newman, Vanderbilt University Medical Center, Nashville, Tennessee 37232, USA.