Article Text

Abstract
Aims: A major breakthrough in the molecular genetics of hypertrophic cardiomyopathy (HCM) has made genetic testing now available in clinical practice, raising new questions about its implications, potential benefits, and the organisation of the procedure. The aim of this work was (1) to discuss the different questions related to genetic testing in HCM, and propose guidelines for the different situations, (2) to report our preliminary experience with a specific procedure.
Methods and results: The main questions asked by patients and relatives concern presymptomatic diagnosis and prenatal counselling/diagnosis, while clinicians sometimes discuss diagnostic and prognostic testing. To take into account the complex medical and psychological implications of this new approach, we developed a specific, multidisciplinary, and multiple step procedure, including a cardiologist, a geneticist, and a psychologist. Seventy subjects were examined, including (1) 29 adults for presymptomatic diagnosis (of whom 10 left the procedure after the first visit and 19 continued, among whom six had a mutation and two experienced negative psychological impact, observed during follow up), (2) nine couples of parents for presymptomatic diagnosis in their children (the procedure was stopped after the first visit in eight and continued in one), (3) 22 couples for prenatal counselling (no prenatal genetic testing was asked for after the first visit), and (4) 10 subjects for diagnostic testing. We decided to perform no prognostic testing.
Conclusion: Our preliminary experience confirms the complexity of the situation and suggests the necessity for a specific procedure to ensure good practice in genetic testing of HCM.
- genetic counselling
- hypertrophic cardiomyopathy
- presymptomatic diagnosis
- prenatal diagnosis
Statistics from Altmetric.com
Hypertrophic cardiomyopathy (HCM) is characterised by unexplained hypertrophy of the left ventricle and remains a leading cause of sudden death in teenagers and young adults, especially in athletes.1–4 The disease is usually genetically transmitted with an autosomal dominant mode of inheritance and considerable progress has been accomplished in the understanding of the molecular genetics of familial HCM over the last decade.5,6 Apart from fundamental and clinical research, new questions are now being addressed to cardiologists and geneticists by patients and relatives about the availability of genetic testing in clinical practice. Equally important for health care providers is the understanding of the basic genetic principles underlying the genetic testing process and also the complexity of psychological effects of genetic testing for individual subjects and their family.7–10 Because of the uncertainties of the medical benefit of genetic testing in HCM, and because of its possible adverse psychological effects, careful consideration is necessary. This has led the French Network on HCM to examine the different situations and to consider the best way to give the genetic results to individual applicants. The aim of the present paper was to discuss the complexity of questions and problems related to genetic testing in HCM, and to report our preliminary experience with a systematic multidisciplinary approach.
THE APPLICANT, GENETIC TESTING, AND THE GENETIC COUNSELLOR
Genetic testing can theoretically be performed in different situations, such as diagnostic testing, prognostic stratification, prenatal diagnosis, and presymptomatic diagnosis, all of which are associated with a variable degree of complexity and psychological implications. The usefulness of the process should be analysed from two different and complementary points of view: that of the clinician who must weigh up the medical benefit and that of the individual applicant whose goal is more related to a “right to know”. In order to ensure the good practice of genetic testing in HCM, the French Network on HCM (members listed in the Appendix) proposed a template and guidelines for genetic counselling. The procedure and proposals were based in part on previous recommendations from genetic societies outside the cardiovascular field11–13 or from cardiological societies.14,15 We proposed that information about genetic testing in HCM should be given in most cases following a multidisciplinary approach by a team made up of a cardiologist, a geneticist, and a psychologist. The three clinicians had consecutive interviews with the applicant in the same place and during the same outpatient visit. The temporal dimension of the counselling was an additional key point. In situations such as presymptomatic diagnosis, a multiple step procedure was mandatory (fig 1) and included the following phases: (1) information about the test, (2) delay before the decision made by the applicant, (3) blood sampling and molecular characterisation, (4) announcement of the result, and (5) follow up by regular interviews to assess the impact of the procedure on the well being of the subject (comparison with the pretesting status by psychological scales), with psychological support when necessary. The first outpatient clinic (with PC, DH, and MG) was created in 1999 at the Pitié-Salpêtriére Hospital, Paris. Over a 30 month period, 70 subjects were seen at this hospital following the multidisciplinary approach (for diagnostic testing, the procedure was simplified).
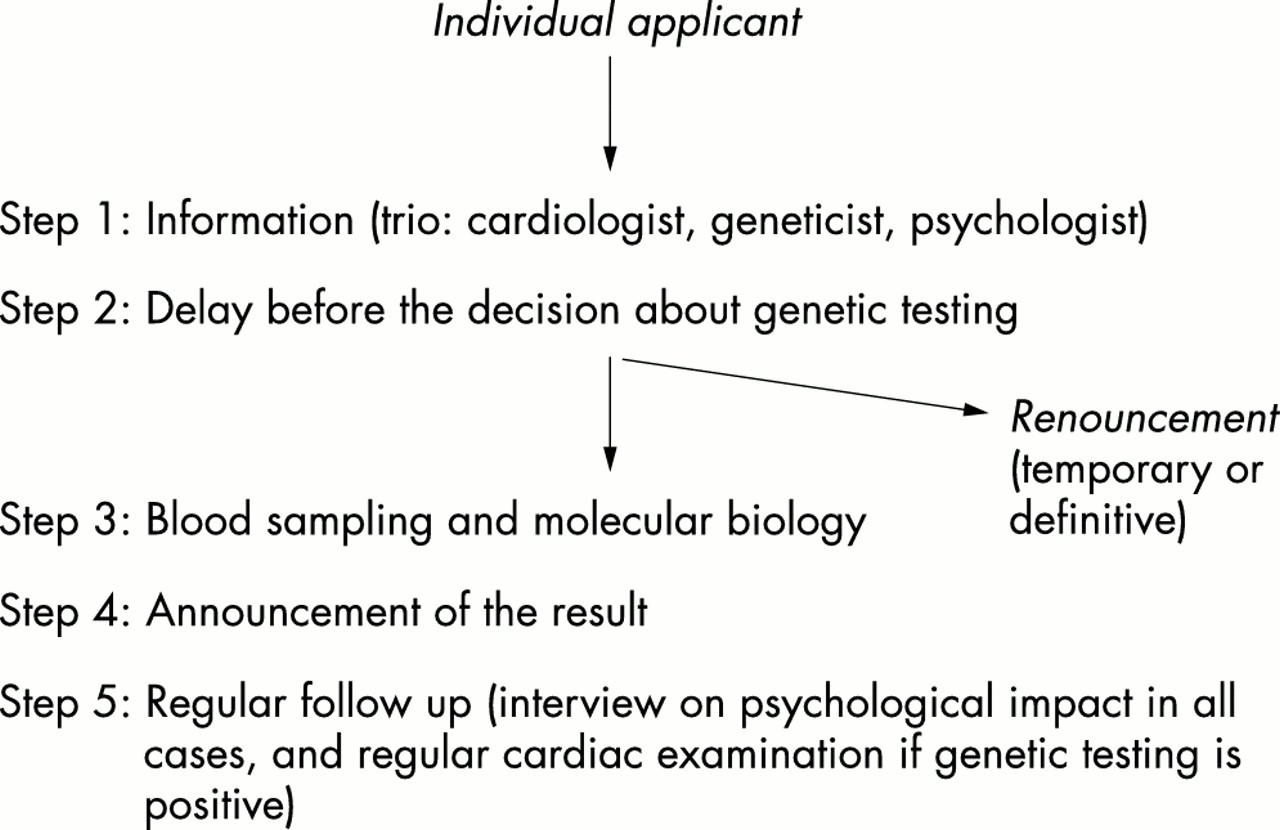
{kind=link}
Procedure for presymptomatic diagnosis in the French Network.
When genetic testing was decided on, then blood samples were taken and sent to the Biochemistry Department for molecular analysis. Since the disease is genetically heterogeneous,5,6 the first molecular step is to identify the mutation responsible for the disease in the index patient in the family. A systematic screening of most recognised genes is performed. Although both time consuming and very costly, the identification of the responsible mutation is possible in the majority of cases, especially since the two most frequent genes account for about 85% of all genotyped families.16 Moreover, the development of more powerful and automated technologies, such as DNA chips or microarrays, will probably greatly modify molecular strategy in the near future.
PRESYMPTOMATIC DIAGNOSIS
In this situation, the applicant is clinically healthy (with a normal echocardiography and a normal ECG), belongs to a family with HCM, and wants to know if he/she has a mutation in the gene responsible for the disease in the family. As a first approach, the theoretical risk of being a healthy carrier is 50% (an a priori probability) when the subject is a first degree relative of a patient with HCM. As penetrance is incomplete (70% in adults) but increases with age, and since cardiac examination is normal in this adult, according to Bayes’s theorem the probability of being a healthy carrier is in fact lower in this situation (an a posteriori probability of about 25%, if the penetrance is 70% in adults) (17). However, genetic testing can give a definitive answer to the question. To a certain extent, clinical tools such as ECG and echography (conventional and tissue doppler) can be considered as a presymptomatic diagnosis,17,18 but with different medical and therapeutic implications. The reason why a subject asks for the test is usually twofold: (1) the psychological burden related to uncertainties about his/her risk of being a carrier of the disease mutation and therefore the risk of developing the disease later, and (2) the wish to know the risk of transmission of the disease to offspring.
The medical benefits of the procedure appear uncertain in this situation. Firstly, limited data are as yet available about the natural history of healthy carriers. The risk of developing the disease later (with obvious hypertrophy), however, appears very high since transverse studies indicate an increasing penetrance with age.17 In contrast, the risk of sudden death in healthy carriers (before the occurrence of left ventricular hypertrophy) is assumed to be very low since such cases have been published in only one family.19 Secondly, no medical treatment is effective in preventing or in lowering the occurrence of the disease. Moreover, there is no evidence that any medical treatment affects prognosis in patients with hypertrophy.2,3,20 The effect of amiodarone against sudden death is controversial. Restriction of physical activity might be recommended but its effect against sudden death in healthy carriers remains speculative. The only treatment that might prevent sudden death is an implantable defibrillator,21 which is proposed for patients with HCM at high risk for sudden death and is not at the moment recommended for healthy carriers. However, this proposal could be questioned in healthy carriers in the context of a family with several premature sudden deaths related to HCM. Thirdly, the expression of the disease is highly variable and the clinician cannot predict the age at onset of the disease nor its severity, although phenotype-genotype correlations may be of some help. Finally, predictive genetic testing may result in adverse psychological consequences. The previous psychological burden because of uncertainty related to the genetic status could then be replaced by a new psychological burden about the near certainty of developing the disease later, including the risk of sudden death.
For these reasons it is not possible to recommend systematic presymptomatic genetic testing, from a medical point of view. If the test is positive, however, strict medical follow up is required which will allow the occurrence of LVH to be diagnosed very early and could lead to improved therapeutic management. The early diagnosis of the expression of the disease will lead to early complementary investigations (such as Holter-ECG and exercise test) which will allow the early identification of subjects at high risk for sudden death and therefore the need for specific treatment. We also recommend restriction of physical activity in genetic carriers, even before the occurrence of hypertrophy (although of uncertain efficacy at this stage, with no medical consensus). If the test is negative, the applicant will be reassured and the procedure will probably result in an improvement of the well being of the subject. The reduction in follow up could also lead to an economic benefit. Our procedure for presymptomatic diagnosis was established as indicated in fig 1. Briefly, it includes a written protocol for the conduct of the testing programme; the fact that the subject or family should be counselled in advance regarding the potential benefit but also the limitations of the test and advised that they had the right not to undergo the test or know the result; the initial counselling should be separated in time (a day or more) from a biological sample being taken; the decision to take the test is solely the choice of the person concerned; written informed consent is signed; molecular biology should be performed according to rigorous standards of accuracy in recognised laboratories; confidentiality of the procedure should be respected so that the person should not be discriminated against in any way (in employment or insurance) as a result of genetic testing.
Twenty-nine adults, all with a mutation previously identified in their family, were seen at the Pitié-Salpêtriére Hospital according to the established procedure (table 1). There were 14 men and 15 women (mean age 37 years, range 18 to 66 years). All were first degree relatives of a patient with HCM, except for three second degree relatives. At least one major clinical event was present in the family of 11 of the applicants. After the first step of information, eight chose to leave the procedure and blood sampling was therefore not performed. Most of them concluded that they would rather not know the information. Two declined because they felt they had the mutation and thus considered the genetic testing unnecessary. Among the 19 subjects who chose to take part in the test, six had the mutation. No major adverse psychological effects were observed after the procedure, with a mean follow up of eight months, except for a subdepressive syndrome in one woman (with a normal psychological condition during the initial procedure) five weeks after the result. In addition, mild hypochondriac symptoms (palpitations and chest pain) appeared in another subject after the result. Thirteen subjects did not have the mutation and were reassured. In one young woman (27 years old), however, the negative result was followed by a paradoxical adverse psychological effect with increased anxiety. Since all the other members of the family had HCM or carried the mutation, she concluded that her father was possibly not her biological father (although she had understood the mode of inheritance of the disease) and she asked us to perform a paternity test (which was declined).
Presymptomatic diagnosis in adults
PRESYMPTOMATIC TESTING IN CHILDREN
Genetic testing in children remains controversial, since the medical benefit of presymptomatic diagnosis in HCM is questionable and since the request is made by the parents and the child is often not mature enough to understand the consequences of the test and make any decisions. Some clinicians have emphasised the potential benefit of testing in HCM,22,23 especially since sudden death may be the first symptom of the disease and it may occur during childhood and adolescence. However, as in adults, the only medical implication is a regular follow up in genetic carriers which will allow the clinical expression of the disease to be diagnosed very early. Other clinicians consider presymptomatic testing in children more deleterious than beneficial,23,24 because of the lack of an efficient treatment to prevent sudden death in a clinically healthy child bearing a mutation, combined with possible adverse psychological effects (such as increased anxiety in parents, possible deleterious consequences on parent-child relations including overprotectiveness, and on the evolving personality of the child). The situation has been compared to some late onset genetic diseases such as Huntington’s disease where there is a consensus to consider such testing as unethical.11,25
Therefore, our Network has reservations about genetic testing in children. An alternative strategy is to propose regular cardiological examination in all children who are first degree relatives, until hypertrophy appears or until the child reaches the age and maturity to make his/her decision. Genetic testing may be performed, however, in selected cases, especially when making a decision regarding the participation of a child in competitive sports. Therefore, a careful case by case approach is mandatory.
Nine couples in genotyped families asked us for predictive genetic testing in their children (n=13, mean age 10 years, range 15 months to 16 years) (table 2). Major clinical events were reported in the family of five couples but without sudden death in the sib or parents of the children involved in the test. After the first step, eight couples of parents did not continue the procedure and asked for time to think about the situation. Genetic testing was performed in the last family in a girl who was 16 years old and wanted to perform the procedure because of sporting competition (riding and dance). In addition, the cardiologist of the family reported a very mild and non-specific abnormality on echocardiography (a normal cardiological examination was, however, found in our institution). She carried the mutation and competitive sport was stopped. A transient panic attack was observed a few days after the result.
Presymptomatic diagnosis in children
GENETIC COUNSELLING AND PRENATAL DIAGNOSIS
The situation is typically that of a couple in a family with HCM who requests information about the risk of transmitting the disease. The theoretical risk of transmitting the disease gene (50% in each pregnancy when one of the two parents is affected by HCM) is explained. However, if the mutation responsible for the disease is identified in the family, then it will be possible to determine if the fetus is a carrier of the mutation or not, through amniocentesis or chorionic villus sampling as early as 10 weeks after the beginning of pregnancy. Since the procedure can result in accidental fetal loss (in about 1%), it is considered only if the couple wishes to draw a practical conclusion from the result, that is, pregnancy termination if the fetus carries the mutation.26
Can the severity of the disease justify medical abortion? From a medical point of view, the severity of HCM is obvious in the case of malignant forms of the disease, with a familial history of sudden deaths. However, this situation is uncommon. The risk of major complications (sudden death or death caused by congestive heart failure) is low (about 1% per year) in the majority of patients with HCM.2,3 In addition, there is such a great variability in the expression of the disease (age at onset of symptoms, severity of symptoms, risk of complication, etc), even within a given family, that a cardiologist cannot precisely predict the outcome of a subject with a mutation.
We therefore provide detailed information not only on the theoretical possibility of prenatal diagnosis but also on the uncertainties of the medical benefit of the procedure. No encouragement to prenatal testing was made during the counselling.
Twenty-two couples were seen for prenatal counselling (table 3). For 15 of them, the mutation was previously identified in the family, and major complications were reported in 10 out of the 15 families. Five of the 22 women were pregnant at the time of the genetic counselling. After the first step of information according to the multidisciplinary approach, none of the couples still requested prenatal diagnosis. Some couples explained that they did not want to have a child because of the high risk, and others that they wanted to have a child whatever the consequences. The five pregnancies continuing at the time of the counselling went to term (three in genotyped families). Six additional pregnancies were reported after the genetic counselling, including two in a genotyped family with several previous major events (two sudden deaths and two cardiac transplantations) and none of the couples requested prenatal diagnosis.
Prenatal counselling/diagnosis
DIAGNOSTIC TESTING
HCM or another cause of left ventricular hypertrophy?
Differential diagnosis is a possible but uncommon indication for genetic testing in HCM. The disease is usually quite easy to distinguish from other causes of left ventricular hypertrophy (LVH). In some cases it might be more difficult, especially in the context of athletes with borderline abnormalities. Here it is crucial to differentiate between physiological hypertrophy and HCM. Since the process of molecular screening is particularly long, costly, and uncertain, our Network decided not to perform genetic testing in these situations, except when a familial context was present.
In our experience, genetic testing was performed for differential diagnosis in only one family with a particular situation. A 13 year old girl experienced cardiac arrest with successful resuscitation. She had no history of cardiac disease and subsequent examination found no cardiac abnormalities, including LVH, except highly permeable atrioventricular node. As there were several subjects with HCM in her family, the cardiologist asked us if cardiac arrest could be related to familial HCM. Although very uncommon, sudden death related to HCM in subjects without LVH has been reported in one family.19 We therefore performed a genetic test in the index patient with HCM. A mutation in the MYH7 gene was identified in the family, but the mutation was absent in the girl. A complete screening of six additional genes was performed and no additional mutation was identified in the family. We therefore concluded that the cardiac arrest was unlikely to be related to the familial HCM.
Sporadic HCM or familial HCM?
Some adults with apparently sporadic HCM asked us if the disease was genetic or not, to determine if there was a risk of transmission of the disease. A systematic cardiological study in first degree relatives (with ECG and echocardiography) is first required to determine if the disease is familial or not. Even when the family study is negative, HCM may have a genetic cause since several de novo mutations have been described.27 According to our procedure, apparently sporadic cases are therefore informed that the disease may be genetic. Nine procedures with molecular analysis are under way and the responsible mutation has already been identified in four cases.
PROGNOSTIC STRATIFICATION
Genetic testing might be useful for the cardiologist to help stratify the risk of a patient with HCM.28 Firstly, conventional cardiological tools, such as Holter-ECG and blood pressure during exercise, have a good negative predictive value (about 95%) for sudden death, but a very poor positive predictive value (about 15%),2,3,20 so that the evolution of the disease is very difficult to predict in a given patient. Secondly, preliminary phenotype-genotype correlations indicate that genetic heterogeneity could account at least in part for the phenotypic heterogeneity.28 A more accurate identification of subjects at high risk of sudden death, using this approach, could lead to prophylactic implantation of a cardioverter defibrillator.21 However, caution is required because available studies are based on a small population (in part because of the low rate of recurrence of a given mutation).28 Further studies based on larger populations are needed to confirm the correlations.
For these reasons, and at least for the present, we consider genetic testing as premature and do not recommend it for prognostic stratification in clinical practice. No genetic testing was therefore performed in this specific situation.
CONCLUSIONS AND PERSPECTIVES
We propose here the first detailed description of the different situations related to genetic testing in HCM, and the first report of a preliminary experience with genetic counselling according to a specific procedure. We used a multidisciplinary approach and a multiple step procedure, based on the wide experience of genetic counsellors in other non-cardiological diseases.7–11 We also propose guidelines for the different situations we met. These proposals express our position. It is however possible that different conclusions would have been made in other countries, owing to differences in the specific culture and the socioeconomic and legal aspects of each country. In addition, the answers will evolve with time, with the increasing progress of molecular technology and of the clinical knowledge of the disease. In any case, the ultimate goal should be an improvement in the quality of life of subjects and their families, without adverse psychological effects, as has been achieved in other genetic diseases thanks to strict and accurate guidelines.8–11
APPENDIX
Coordinators of the French Network on Hypertrophic Cardiomyopathy are M Komajda and K Schwartz. Members are: L Carrier, K Schwartz (INSERM U523, Hôpital Salpêtriére, Paris); P Richard, C Ledeuil, B Hainque (Service de Biochimie, Hôpital Salpêtriére, Paris); P Charron, A Bénaïche, R Isnard, M Komajda (Service de Cardiologie, Hôpital Salpêtriére, Paris); D Héron, M Gargiulo (Département de Génétique, Hôpital Salpêtriére, Paris); J F Forissier, O Dubourg (Service de Cardiologie, Hôpital A Paré); A Hagége, M Desnos (Service de Cardiologie, Hôpital Européen Georges Pompidou, Paris); J M Langlard, J B Bouhour (Hôpital Laennec, Nantes); A Millaire (Hôpital Cardiologique, Lille); J Feingold (INSERM U155, Lonchamps, Paris); L Duboscq-Bidot; E Villard (Ass Claude Bernard et Université Paris VI, Hôpital Salpêtriére, Paris).
Acknowledgments
This work was supported by INSERM (Réseau de Recherche Clinique No 4R009B), the Association Française contre les Myopathies, the Fédération Française de Cardiologie, and the Délégation à la Recherche Clinique de l’Assistance Publique-Hopitaux de Paris (Crédits EMUL et IFR Coeur, Muscle et Vaisseaux).