Article Text

Abstract
Pulmonary lymphangioleiomyomatosis (LAM) is a rare disorder limited almost exclusively to women of reproductive age. LAM affects about 5% of women with tuberous sclerosis complex (TSC). LAM also occurs in women who do not have TSC (sporadic LAM). TSC is a tumour suppressor gene syndrome characterised by seizures, mental retardation, and tumours in the brain, heart, and kidney. Angiomyolipomas, which are benign tumours with smooth muscle, fat, and dysplastic vascular components, are the most common renal tumour in TSC. Renal angiomyolipomas also occur in 63% of sporadic LAM patients. We recently found that 54% of these angiomyolipomas haveTSC2 loss of heterozygosity, leading to the hypothesis that sporadic LAM is genetically related to TSC. In this study, we screened DNA from 21 women with sporadic LAM for mutations in all 41 exons of TSC2. Twelve of the patients had known renal angiomyolipomas. No TSC2mutations were detected. We did find three silentTSC2 polymorphisms. We conclude that patients with sporadic LAM, including those with renal angiomyolipomas, do not have a high frequency of germline mutations in the coding region of TSC2.
- TSC2
- pulmonary lymphangioleiomyomatosis
Statistics from Altmetric.com
Pulmonary lymphangioleiomyomatosis (LAM) is a rare and often fatal disease of unknown aetiology affecting almost exclusively young women.1 2 Microscopically, LAM is characterised by diffuse infiltration of the pulmonary interstitium by smooth muscle cells and cystic distortion of the lung architecture. LAM can occur as an isolated disorder (sporadic LAM) or in association with tuberous sclerosis (TSC). TSC is an autosomal dominant disorder characterised by seizures, mental retardation, and hamartomatous tumours of the brain, heart, kidney, lung, and skin. LAM affects 2.3% of people (or 4.6% of women) with tuberous sclerosis.3 Renal angiomyolipomas, which are benign tumours composed of fat, smooth muscle, and dysmorphic vessels, occur in 70% of TSC patients and in 63% of women with sporadic LAM.4-6 The occurrence of angiomyolipomas in both TSC associated and sporadic LAM has led to the hypothesis that these diseases have a common genetic basis.4 7
There are two TSC genes, TSC1 on chromosome 9q348 and TSC2 on chromosome 16p13.9 The frequency of TSC1or TSC2 loss of heterozygosity (LOH) in TSC associated angiomyolipomas is approximately 60%.10 In contrast, TSC2 LOH occurs in only 10% of angiomyolipomas from patients who do not have either TSC or LAM.11 We recently found that 54% of angiomyolipomas from women with sporadic LAM have LOH in the TSC2region of chromosome 16p13.12 We did not findTSC1 LOH in the sporadic LAM associated angiomyolipomas.12
The TSC2 LOH in the LAM associated angiomyolipomas led us to test the hypothesis that some patients with sporadic LAM have de novo germline mutations inTSC2 associated with a mild phenotype affecting only lung and kidney. Phenotypic variability in TSC is common, with some patients having only mild signs or symptoms of disease, and approximately 70% of TSC patients have new germline mutations, with no previous family history of the disease. We therefore analysed DNA from 21 women with sporadic LAM forTSC2 mutations. The sources of DNA were peripheral blood lymphocytes (12 patients), cultured lung cells obtained at the time of lung transplantation (eight patients), and angiomyolipoma (one patient).
Patients, methods, and results
This study was approved by the Institutional Review Board of Fox Chase Cancer Center. None of the patients in this study had dermatological or neurological signs or symptoms of TSC. Twelve of the patients had known angiomyolipomas (table 1). The angiomyolipoma from patient 492 had TSC2LOH.12 The angiomyolipomas from patients 423, 480, 481, 489, and 491 did not have TSC1 orTSC2 LOH.12 Tissue from the remaining six angiomyolipomas was not available for LOH analysis. The source of DNA was either peripheral blood lymphocytes or a lymphoblastoid cell line for 12 patients (table 1). For one patient (423) DNA was isolated directly from fresh angiomyolipoma tissue. Fresh or frozen angiomyolipoma tissue from which genomic DNA could be prepared was not available from the other patients. For eight patients, DNA was isolated from primary cultures of lung tissue established at the time of lung transplantation for LAM.
Presence or absence of angiomyolipomas in patient, and source of DNA for mutational analysis
Single strand conformation analysis (SSCP) was used to search for mutations in the coding regions of the TSC2gene. The primers amplifying each of the 41 exons ofTSC2 and the PCR conditions have been previously reported by Au et al. 13 The PCR products were run on MDE gels (AT Biochem). To maximise the detection of variant bands, each PCR product was run on two gels: one without glycerol and one with 5% glycerol. Samples in which variant bands were detected were reamplified and sequenced.
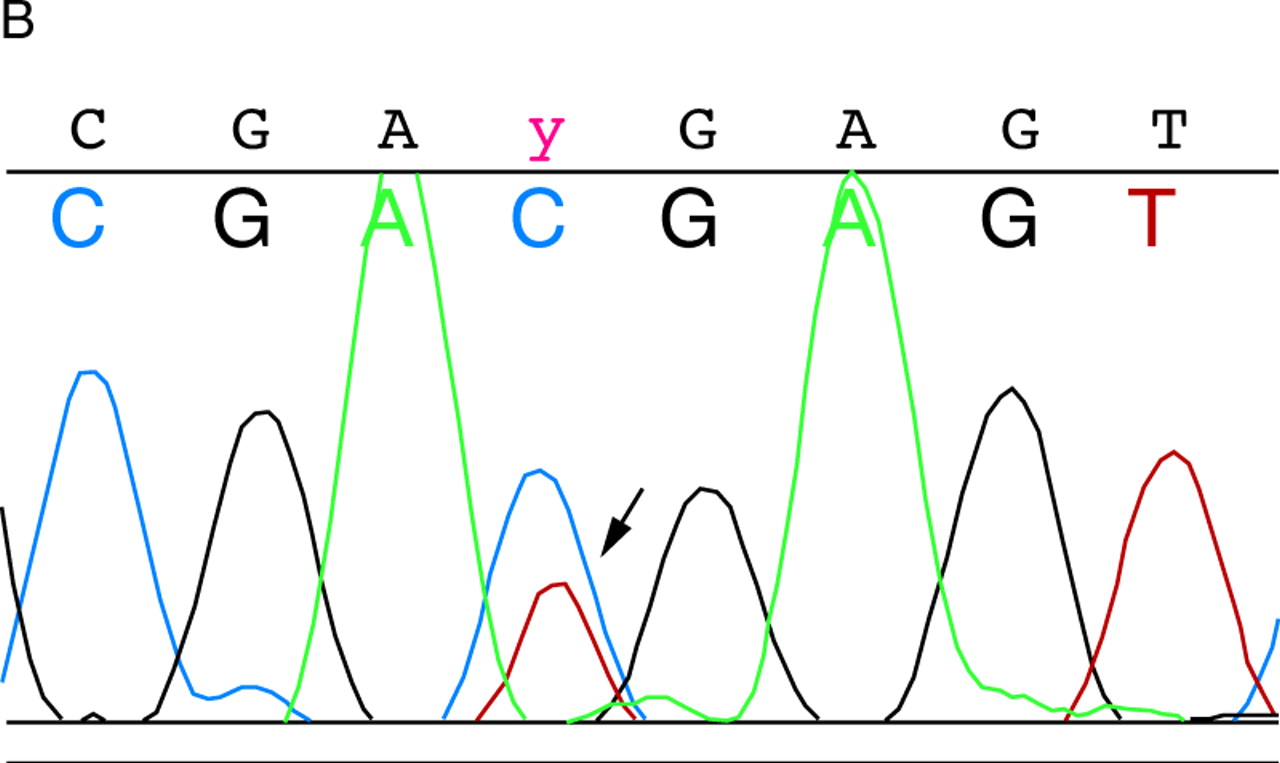
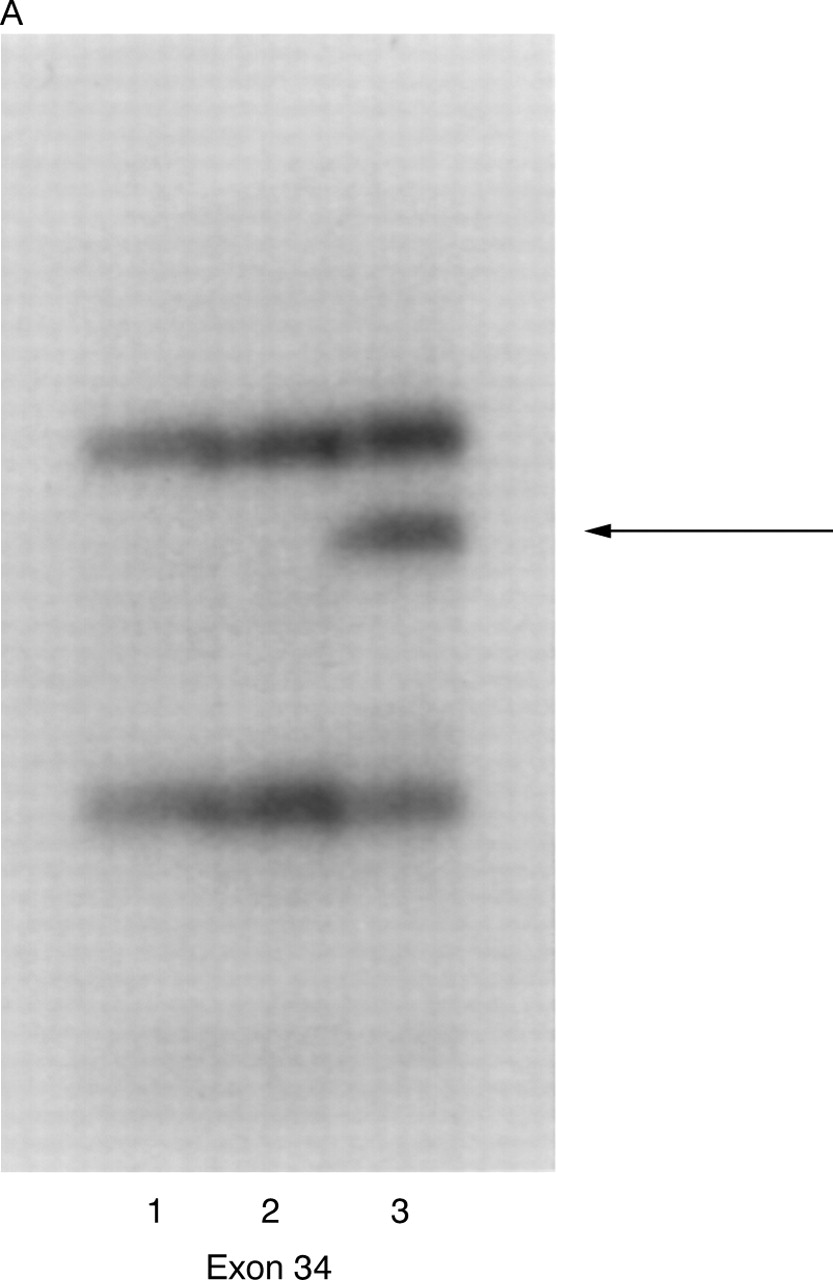
We found three variant bands in exons 12, 14, and 34, all of which were found by sequencing to represent silent polymorphisms (table 2). All three polymorphisms have been previously reported and are present in the TSC Variation Database (http://expmed. bwh.harvard.edu/ts/). NoTSC2 alterations resulting in amino acid changes were detected.
TSC2 polymorphisms in LAM patients
Discussion
The clinical and pathological similarities between TSC and sporadic LAM have led to the hypothesis that LAM is a form of TSC.4 7 Our recent finding that 54% of angiomyolipomas from sporadic LAM patients have LOH in theTSC2 region of chromosome 16p1312 supported this hypothesis. In this study, DNA samples from 21 women with pulmonary LAM, 12 of whom also had renal angiomyolipomas, were examined for TSC2mutations in all 41 exons using SSCP. No definite mutations were identified.
Our data indicate that germline mutations inTSC2 are infrequent in sporadic LAM. In addition, the lack of TSC2 mutations in the eight DNA samples from primary cell cultures established from LAM lung tissue at the time of lung transplantation suggests that somatic mosaicism for TSC2 mutations is not a frequent cause of sporadic LAM. It is possible that in some patients, however, mutations were missed. The sensitivity of SSCP inTSC2 analysis is not known, but for other genes the sensitivity for a single gel condition has been estimated at 75-98%.14 Each PCR product in our study was run under two gel conditions (with and without glycerol). We also did not screen for mutations in the non-coding regions of TSC2or for deletions. Large TSC2 deletions are often associated with renal cysts,15 which were not present in any of the patients in this study. We did not analyse these samples for TSC1 mutations because we had previously found TSC2 LOH, but notTSC1 LOH, in angiomyolipomas from women with sporadic LAM.12
In summary, this is the first report of TSC2mutational analysis in patients with pulmonary lymphangiomyomatosis. We did not find TSC2 mutations in DNA isolated from lymphocytes (12 patients), primary cultures of lung tissue (eight patients), or angiomyolipoma (one patient). We conclude that germline mutations in the coding regions of TSC2 are uncommon in sporadic LAM despite the striking clinical and pathological overlap between these two diseases. Additional studies will be required to determine whether other types of TSC2mutations not detected by SSCP occur in sporadic LAM, whether some patients have somatic mosaicism for TSC2mutations, or whether mutations in other genes are involved.
Acknowledgments
We are grateful to William A Petri Sr and Drs Ann Petri, Becky Raftogianis, and Warren Kruger for critical review of the manuscript, and to Dr Alfred Knudson, Dr Frank McCormack, Ms Sue Byrnes, and The LAM Foundation for their ongoing support. We also thank the patients who contributed blood and tissue specimens for this research. This work was supported by The LAM Foundation (Cincinnati, OH) and the National Institutes of Health (HL 60746).
References
{kind=link}
{kind=link}
(A) Detection of variant band in exon 34. Lanes 1 and 2 contain DNA from controls and lane 3 contains DNA from patient 539. The arrow indicates the variant band. (B) Sequencing of exon 34 from patient 539. The arrow indicates the double peak of C and T at nucleotide 4536.