Article Text

Abstract
Objective: A retrospective study of the UK Pulmonary Hypertension Service for Children for the first 5-year period of its existence.
Design and patients: Records of 216 children with idiopathic pulmonary arterial hypertension (IPAH) and associated pulmonary arterial hypertension (APAH) were reviewed. Kaplan-Meier survival curves were constructed for different diagnostic groups and for different therapies.
Results: At cardiac catheterisation only 7.4% of those with IPAH and 6% of those with APAH responded positively to vasodilator testing and so were treated with nifedipine. Others needing treatment were given continuous intravenous epoprostenol, bosentan or sildenafil singly or in combination. For IPAH survival rates were 85.6%, 79.9% and 71.9% at 1, 3 and 5 years, respectively, compared with a survival time of less than a year in historical untreated controls. A combination of intravenous epoprostenol with either bosentan or sildenafil, or both, appeared to achieve the best outcome. Six children underwent lung transplantation. In APAH survival rates were 92.3%, 83.8% and 56.9% at 1, 3 and 5 years, respectively, postoperative congenital heart disease with severe pulmonary hypertension having the worst outcome.
Conclusion: New pulmonary hypertension-specific medicines have improved survival in children as in adults. Outcome in this series compares favourably with international outcome data.
Statistics from Altmetric.com
Sustained pulmonary arterial hypertension is associated with the development of pulmonary vascular disease, which when advanced leads to right heart failure and death. Pulmonary arterial hypertension is subdivided into idiopathic pulmonary arterial hypertension (IPAH) and associated pulmonary arterial hypertension (APAH) when present with other disorders.1 Left untreated, children with IPAH fare less well than adults. The predicted survival after diagnosis is less than a year compared with 2.8 years in adults.2 The introduction of pulmonary hypertension specific therapies has improved the prognosis in both adults and children. Before the definitive trial of intravenous epoprostenol in adults in 1996 the only medications available were calcium channel antagonists, which are efficacious in only a minority. The beneficial effect of epoprostenol in children with IPAH was demonstrated three years later.3 Anticoagulation was, and is, also considered helpful in adults with IPAH.4 5 The oral drug bosentan, a dual endothelin receptor antagonist, was found to be efficacious in adults in 2002,6 and later we and others showed that it was also efficacious in children.7 8 The phosphodiesterase V (PDE-5) inhibitor sildenafil was shown to be efficacious in a 12-week trial in adults with IPAH.9 A trial is in progress to study the effect of the drug in children.
We established a clinical network for the care of all children with significant pulmonary hypertension in the United Kingdom to try to optimise the treatment of this patient population using new and emerging therapies. This network was designated by the National Specialist Advisory Group of the Department of Health in 2002. It was commissioned as a funded service by the Specialist Commissioning Group in April 2007.
The UK Pulmonary Hypertension Service for Children was organised on a hub-and-spoke principle, the hub being at Great Ormond Street Hospital for Children (GOSHC). The spokes were six major centres of paediatric cardiology in the UK—namely, the Freeman Hospital, Newcastle upon Tyne, Leeds General Infirmary, Bristol Children’s Hospital, Birmingham Children’s Hospital, Yorkhill Hospital, Glasgow and the Royal Belfast Children’s Hospital. Joint clinics were held at these hospitals by SGH with the local paediatric cardiologist. We hope that all children with IPAH are now referred to the service. Children who are referred with APAH generally have severe pulmonary hypertension. Cases of mild PAH are not usually referred to the service. Children with mild postoperative PAH, for example, are usually managed locally by their own physicians. Pulmonary hypertension within the first month of life is specifically excluded from the service, with few exceptions such as a documented or suspected family history of IPAH.
We present a retrospective analysis of the treatment given and the response to treatment in terms of survival in children treated by this service for the first 5-year period.
METHODS
A total of 216 patients with either IPAH or APAH were treated between April 2001 and March 2006. All children were assessed initially by clinical examination, ECG, chest x-ray and echocardiography, to confirm the diagnosis and determine the degree of right ventricular impairment. Children over five years of age carried out a 6-minute walk test. Cardiac catheterisation was performed in 146 children, 90% of those with IPAH and 53% of those with APAH. Ten per cent of children with IPAH were considered too ill to undergo cardiac catheterisation. A recent publication details the management of children with PAH.10
Initial treatment
IPAH
The treatment algorithm used was based on the European Society of Cardiology algorithm for the management of patients with IPAH and the national guidelines.11 12 The few children with a positive response to acute vasodilator testing with nitric oxide at cardiac catheterisation were given a calcium channel antagonist, nifedipine. Those who did not respond were treated initially either with the oral drug bosentan or, if more symptomatic and in WHO class III or class IV functional status, intravenous epoprostenol. The therapeutic regimen was tailored to the needs of each child and adjusted according to response. Children receiving the dual endothelin receptor antagonist bosentan (Tracleer) had a target dose of 31.5–125 mg twice a day, according to weight.Since the principal side effect of this drug is elevation of liver enzymes, liver function was checked before instituting therapy and at monthly intervals while on therapy. For the first month of treatment the dose was half the target dose. The dose of sildenafil was 0.5–1.5 mg/kg, rarely more, according to symptomatology. The starting dose of intravenous epoprostenol was 2 ng/kg/min and increased until a satisfactory clinical response was obtained, up to 60 ng/kg/min or more in some children. The principal side effects of this drug in children are diarrhoea on instituting or intensifying therapy and jaw discomfort. The majority of children were anticoagulated with warfarin but the small children received aspirin.
An atrial septostomy was performed at the initial catheterisation study in children who presented with syncope.
APAH
The treatment regimen depended on the underlying condition, symptomatology and the findings at cardiac catheterisation. Those with postoperative pulmonary hypertension, HIV and connective tissue disease who had a pulmonary arterial pressure at or approaching systemic level and a high resistance were treated using the same rationale and the European Society of Cardiology algorithm as were children with IPAH. Symptomatic children with Eisenmenger syndrome and chronic lung disease were given monotherapy, the drug chosen depending on age, sex and acceptance of the blood tests mandatory with the use of bosentan.
Follow-up of IPAH and APAH
The maximum follow-up for the total population was 5 years, minimum 1 month, with a median of 1.79 years. Children were assessed every 1–4 months to monitor the response to treatment, to carry out further investigations and modify therapy as necessary. Routine investigations included an ECG, echocardiographic study and a 6-minute walk test when feasible. Repeat cardiac catheterisation studies were carried out when clinically indicated and not as a routine.
Statistical methods
Patient age, haemodynamic data and WHO functional class are expressed as mean and standard deviation. The median and range are also given. Diagnostic groups were compared using one-way ANOVA and Pearson correlation. Survival was calculated using Kaplan-Meier analysis (SPSS) starting from April 2001 for those already diagnosed with PAH and from the time of first presentation for all those presenting after this date. For all survival estimates patients were censored at the time of transplantation and at the end date of the analysis, 31 March 2006.
Survival curves were determined for: (1) all cases, (2) all cases of IPAH and APAH, (3) specific subgroups of APAH, (4) IPAH according to treatment regimen, (5) APAH due to postoperative congenital heart disease (CHD) according to treatment regimen. Survival times are given as the mean (SE).
PATIENT CHARACTERISTICS AT PRESENTATION
Idiopathic pulmonary arterial hypertension
In the 5-year period, 60 children had IPAH (table 1). The female:male ratio was 1.6:1. Twenty-three children had a small atrial communication, generally thought to be a patent foramen ovale. Five had a small incidental ventricular septal defect. Five children had Down syndrome, all two years of age or less at presentation. The mean age at presentation to the service was 7.37 (5.7) years, median 6.34 years. The mean WHO functional class was 3.3 (0.55). All had ECG and echocardiographic evidence of right ventricular hypertrophy. Cardiac catheterisation (54 cases) showed a mean pulmonary artery pressure (PAP) of 61.9 (20.2) mm Hg and a pulmonary vascular resistance (PVR) of 22.01 (10.9) units/m2. Four of these patients (7.4%) showed a fall in resistance to a near normal level when tested with inhaled NO or 100% oxygen in the earliest studies. Six children were not catheterised because they were critically ill on admission. Three have since died.
Associated pulmonary arterial hypertension
A total of 156 cases were investigated as shown in table 2. The mean age at presentation of all the children was 7.89 (5.9), median 7.46 years. The female:male ratio was 1.1:1. They were in WHO functional class 2–4, mean 2.95 (0.61). Cardiac catheterisation (82 cases) showed a mean PAP of 48.8 (16.03) mm Hg and a mean PVR of 15.6 (11.3) units/m2 with a wide range of values. Four patients had a PVR of less than 4 units/m2. Three of these had postoperative pulmonary hypertension and one had complex CHD and has since had a successful intracardiac repair. The mean PAP for each of the six subgroups was between 39.8 mm Hg and 58.4 mm Hg and the mean PVR was between 7.9 units/m2 and 22.7 units/m2 (table 2).
TREATMENT INITIATION AND MAINTENANCE
Choice of therapy was determined by the aetiology of the pulmonary hypertension and the parents’ wishes, and by international guidelines.11 12
Idiopathic pulmonary arterial hypertension
Initiation of treatment
According to the rationale of the European Society of Cardiology algorithm and depending on the child’s clinical condition and the drugs available at the time, 45 children were given monotherapy after diagnosis. The four children with a positive vasodilator response at cardiac catheterisation received nifedipine, the remainder either epoprostenol or bosentan. Thirteen severely sick children were given combination therapy immediately they were referred to the service because they were deteriorating on their current medication. In all children therapy was modified as and when necessary. Two children were not treated; one dying immediately after presentation and one with multiple handicaps whose parents decided should not be treated.
At the close of the study period or at the time of death or transplantation 25 children were on monotherapy and 29 children were on combination therapy (table 1). Twenty-four children required an atrial septostomy for syncope and/or right heart failure.13
Associated pulmonary arterial hypertension
Of the total of 156 children 78% were treated with disease-specific therapies and 22% were not treated (table 2) either because they were relatively asymptomatic or because their parents considered that the side effects and/or mandatory blood tests would have reduced their quality of life. Determinants of choice of therapy are given in the Methods section.
Of the 47 children with postoperative pulmonary hypertension, 40 were on disease modifying therapy, 25 receiving either bosentan or sildenafil, the more severely symptomatic receiving bosentan. Another 11 children received combination therapy. Five of this last group received intravenous epoprostenol: four children were treated with nifedipine alone, seven were untreated. Thus symptomatology and haemodynamics varied widely in the postoperative patients but fewer needed combination therapy than did those with IPAH. Thirty-eight symptomatic children with Eisenmenger syndrome were treated with oral monotherapy, sildenafil or bosentan. One child had been started on nifedipine at another institution, a drug that is not advisable in this condition. Treatment of patients in the other groups depended on disease aetiology and symptomatology.
An atrial septostomy was only carried out in one child with connective tissue disease.
OUTCOME
The survival rates for the entire group of IPAH (60 cases) and APAH (156 cases) were 90.5%, 82.8% and 64.2% at 1, 3 and 5 years, respectively. These figures include the small number of untreated cases. There was no significant difference between the survival rates for IPAH and APAH (fig 1).

Idiopathic pulmonary arterial hypertension
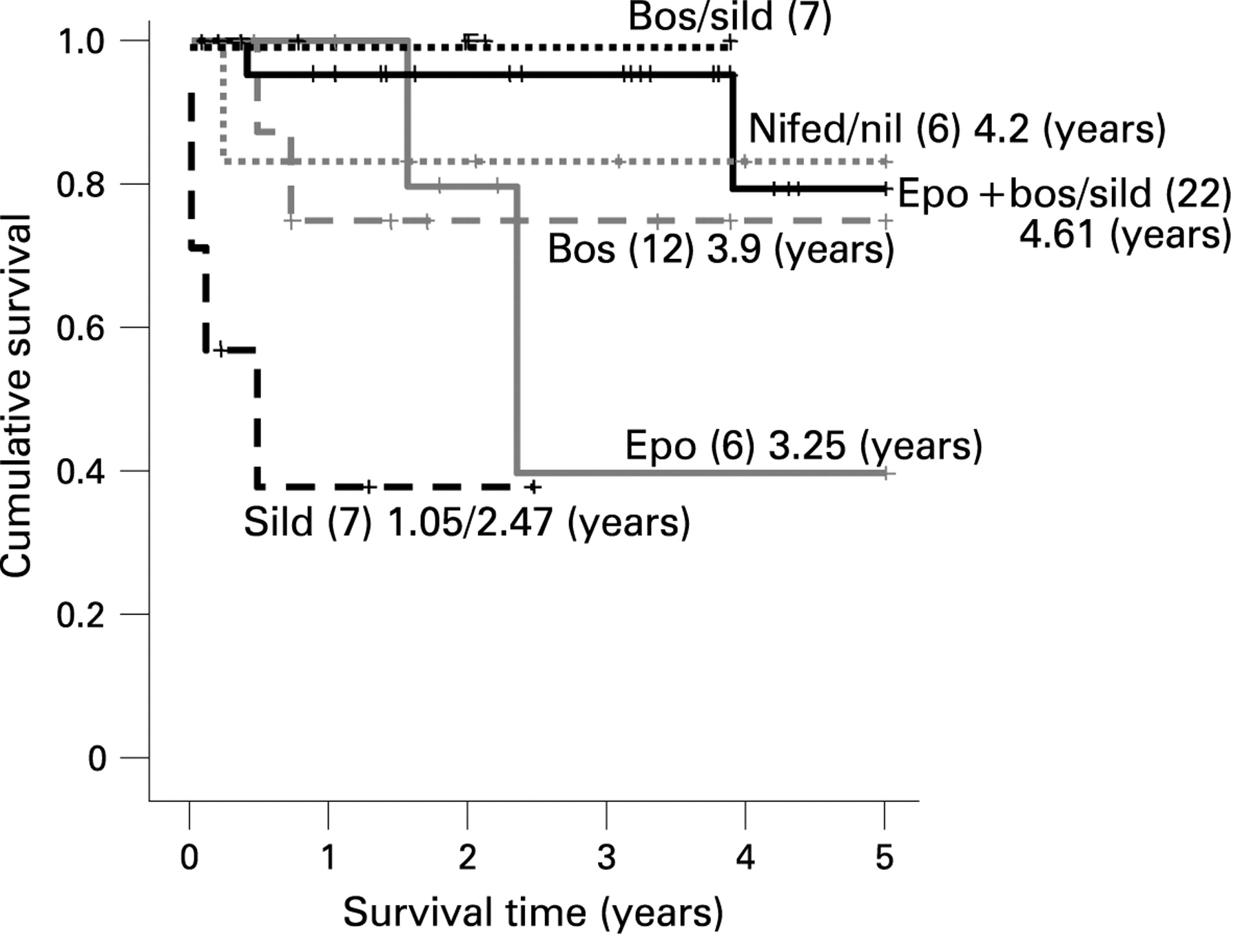
The survival rates at 1, 3 and 5 years were 85.6%, 79.9% and 71.9%, respectively. The mean predicted survival time was 4.07 years (SE 0.25) of the 5-year period assessed (fig 1). Eleven (18%) patients died, eight within one year of presentation. Four were less than 2 years old. Six children had a successful double-lung or heart-lung transplantation. All had been treated with epoprostenol and bosentan for up to 4 years before transplantation. All are alive and well. Four patients diagnosed 6, 7, 8 and 15 years ago are still alive on treatment with either bosentan or nifedipine or epoprostenol (n = 2), respectively.
When survival was analysed according to treatment (fig 2) those on monotherapy with either epoprostenol or bosentan had a predicted survival of 3.25 (SE 0.84) years or 3.9 (SE 0.67) years respectively. The 22 children treated with a combination of epoprostenol and either bosentan or sildenafil or both (five on triple therapy) had a predicted survival of 4.61 (SE 0.25) years of the 5-year period assessed. Six children treated with a combination of bosentan and sildenafil are all alive.
Associated pulmonary arterial hypertension
Survival rates at 1, 3 and 5 years were 92.3%, 83.8% and 56.9%, respectively. The mean predicted survival time for all cases with APAH was 4.06 (SE 0.17) years of the total 5-year period assessed (fig 1). Twenty-three children (14.7%) died. Outcome was dependent on the underlying condition (fig 3). The cumulative survival curves for the subgroups showed, not surprisingly, that those with the best outcome had Eisenmenger syndrome (fig 3). The predicted survival at 5 years was 95.3%. The worst outcome was seen in children with postoperative congenital heart disease, where 11 out of 47 (23%) had died, and in those with connective tissue disease, where three out of nine children had died. The predicted survival at 1 and 3 years was 88.2% and 73% for post operative congenital heart disease and 85.7% and 64.3% for those with connective tissue disease.

{kind=link}
{kind=link}
{kind=link}
In the post-operative cases, bosentan was the most frequently used drug.The predicted survival for this bosentan treated group was 3.67 (SE 0.4) with, out of a possible 4.88 years, 26% of children dying. The combination of bosentan with epoprostenol or sildenafil was associated with a slightly better survival of 4.11 (SE 0.7) years with, out of a possible 5 years, 20% dying. Sildenafil monotherapy gave a predicted survival of 2.21 (SE 0.5) with, out of a possible treatment time of 3.07 years, two out of 10 cases dying.
DISCUSSION
This is the first 5-year report on the UK Pulmonary Hypertension Service for Children. When this national clinical network was established in 2001 the only treatment available for the majority of patients with IPAH was continuous intravenous epoprostenol. Other drugs became available during the course of the 5-year study period. Patients with APAH also benefited as the experience gained in treating IPAH with the new drugs was extended to them. Our survival figures for children with both IPAH and APAH compare favourably with those of other European and American studies. Inevitably, the major limitation of this study is the short follow-up period, the mean time being 1.98 (1.39) years.
It is difficult to compare survival data in different series, particularly in an era when new drugs are being introduced. Most studies have focused on the results of treatment with epoprostenol in IPAH, while others have analysed the short-term response to a new drug. Our survival figures of 85.6% at 1 year, 79.9% at 3 years and 71.9% at 5 years in children with IPAH compare favourably with adult studies on epoprostenol-treated patients which showed 1, 3 and 5-year survival rates of 88%, 63% and 47% in one study14 and 85%, 63% and 55% in another.15 One paediatric study16 reported survival rates of 94%, 88% and 81% at 1, 3, and 5 years, 20.9% of children received a transplant compared with 10% in our series. Treatment success rates in that study, for which death, transplantation and atrial septostomy were censoring events were 83%, 66% and 57% at 1, 3 and 5 years. Thus it would appear that the paediatric survival rates are not very different from those in adults and those in our series are comparable to those in the major studies published in the present era. Children have benefited from new therapies as much as adults, more so when considering that for IPAH the prognosis without therapy is worse in children.
In APAH the underlying diagnosis influences outcome. The survival rates of 92.3%, 83.8% and 56.9% at 1, 3 and 5 years are similar to those in an adult study combining IPAH (72%) and APAH, in which the rates were 93% and 79.9% at 1 and 3 years, respectively.17 Only 53% underwent cardiac catheterisation, primarily those with post-operative PAH, connective tissue disease and the less symptomatic patients with Eisenmenger syndrome. The majority of patients with postoperative pulmonary hypertension referred to the service had severe disease and many fared badly. Seventy-seven per cent received either intravenous epoprostenol, bosentan, sildenafil or a combination of these drugs. Atrial septostomy was not required because the symptomatic children with a high PVR already had an atrial communication. It is well recognised that surgical repair in patients with an elevated pulmonary vascular resistance can accelerate the development of pulmonary vascular disease, whereas those with Eisenmenger syndrome can live untreated for many years.18 In the present study the predicted 5-year survival was better in the children with Eisenmenger Syndrome than in those with postoperative pulmonary hypertension. It should, however, be emphasised that mild pulmonary hypertension can persist for some time after operation and either resolve with time or remain unchanged. Treatment with disease-modifying therapies may only be needed if the child has an additional lesion which puts them at increased risk. These patients are well cared for locally but can be referred to the service if there is evidence of disease progression or any cause for concern. The outcome in the small number of children with connective tissue disease was poor, as it is in adults.19
The children with APAH were treated because they were symptomatic and the drugs had been used to good effect in adults with the same condition such as Eisenmenger syndrome,20 21 connective tissue disease19and interstitial lung disease,22 all treated with bosentan. Infants with chronic lung disease were given sildenafil. There is considerable experience of using this drug to treat pulmonary hypertension in infancy.23
The children with IPAH were treated according to the guidelines current at the time they presented,11 12 24 taking into account the limitations imposed by childhood.
The majority of children received warfarin anticoagulation although the small children received aspirin to try and prevent thrombosis-in-situ. This practice is based on the efficacy demonstrated in IPAH in adults4 but there are no data on children. The main determinant of treatment was, and remains, the response to vasodilator testing with nitric oxide at cardiac catheterisation. Only 7.4% of the children with IPAH who had a catheterisation study showed a positive vasodilator response and so were suitable for treatment with a calcium channel antagonist. Thus far none has relapsed. Until recently the definition of a positive responder has been a fall in pulmonary arterial pressure and resistance of 20% but despite such a fall the PAP and PVR may still be high. We have always used a stricter definition of a positive response. A recent adult European study found that 12.6% of adults with IPAH demonstrated reactivity but only 6.8% had a favourable long-term response to a calcium channel antagonist. On acute vasodilator testing the mean pulmonary arterial pressure was 33 (8) mm Hg in the group with a long-term response to a calcium channel antagonist.25
In our patients with IPAH, 25 of the 47 who were treated with expensive monotherapy initially remained on monotherapy until the end of the study period. Survival was comparable in those given bosentan and epoprostenol, 3.9 years compared with 3.25 years of the 5-year study period, but the clinical status of those given bosentan was better at the time treatment was initiated. We previously reported that bosentan helped improve and stabilise clinical status but epoprostenol was also required in 60% of cases, indicating the need for close monitoring in children treated with bosentan alone.7 These findings were similar to those in a North American study in which half of the patients also needed a prostanoid and 25% a PDE-5 inhibitor.8 Clinical and haemodynamic improvement was maintained in 90% of adults on bosentan monotherapy for more than one year.26 The long-term consequences of starting treatment with bosentan and then adding epoprostenol should deterioration occur are uncertain in children. In adults, however, the addition of epoprostenol improved survival and survival was comparable to that achieved by treating with epoprostenol from the beginning.27 The children with IPAH treated only with epoprostenol appeared to fare worse than adults but no definitive conclusion can be reached based on the small number of children treated. These cases predated the introduction of endothelin receptor antagonists and PDE-5 inhibitors. Outcome was also unsatisfactory in children treated with sildenafil alone. All these patients had been given this drug before transfer to our unit and all were extremely ill on arrival. Given this experience, we have not started any child with IPAH on long-term sildenafil as monotherapy, although it was shown to be effective in adults during a short 12-week study.9 An international multicentre paediatric study is in progress.
In the relatively small number of children studied, the survival rate was better in those children given combination therapy with epoprostenol and bosentan, with or without sildenafil, than in those on monotherapy. The indication for additional therapy was either an unsatisfactory response to initial therapy (epoprostenol or bosentan) or clinical deterioration. The concept of starting treatment with a single agent, preferably an oral drug, followed by the addition of a different agent(s) if necessary is supported by the benefit shown in several adult studies.11 17 28 Initiation of treatment with combination therapy has not been formally evaluated. Combination therapy is a logical therapeutic approach in that it targets different signalling pathways, the prostacyclin, endothelin and nitric oxide pathways, all known to function abnormally in pulmonary hypertension. In giving combination therapy, we did not encounter any problem with drug interaction.29 30
The UK Pulmonary Hypertension Service has provided a structured clinical network to treat children rapidly and to provide all children with immediate access to all new medicines. Lung transplantation is not a satisfactory alternative. The current predicted 50% survival for lung transplant in children is 4.3 years with 75% survival at one year,31 and there are too few donors. New drugs are rarely given to children until proved safe and efficacious in adults, but organised clinical networks provide a framework in which to explore new therapeutic options.
Acknowledgments
We thank the physicians who referred children to the Service and helped care for them. We also thank Yvette Flynn, our clinical specialist nurse, for all her support.
REFERENCES
Footnotes
Funding: AAH was partially funded by Actelion Ltd.
Competing interests: SGH has acted as a consultant and received unrestricted educational grants from Actelion Ltd, Encysive pharmaceuticals, GlaxoSmithKline and Pfizer.
Ethics approval: This work was approved by the ethics committee of Great Ormond Street Hospital and UCL Institute of Child Health.