





Abstract
At any point 1–5 days following ascent to altitudes ≥2500 m, individuals are at risk of developing one of three forms of acute altitude illness: acute mountain sickness, a syndrome of nonspecific symptoms including headache, lassitude, dizziness and nausea; high-altitude cerebral oedema, a potentially fatal illness characterised by ataxia, decreased consciousness and characteristic changes on magnetic resonance imaging; and high-altitude pulmonary oedema, a noncardiogenic form of pulmonary oedema resulting from excessive hypoxic pulmonary vasoconstriction which can be fatal if not recognised and treated promptly. This review provides detailed information about each of these important clinical entities. After reviewing the clinical features, epidemiology and current understanding of the pathophysiology of each disorder, we describe the current pharmacological and nonpharmacological approaches to the prevention and treatment of these diseases.
Abstract
Lack of acclimatisation is the main risk factor for acute altitude illness; descent is the optimal treatment http://ow.ly/45d2305JyZ0
Introduction
Large numbers of people are ascending to high altitudes for the purposes of pleasure, work and athletic competition. Among other important changes, such as decreases in temperature and ambient humidity, the defining environmental feature at high altitude is a drop in barometric pressure, which causes a decrease in the partial pressure of oxygen at every point along the oxygen transport cascade from ambient air to cellular mitochondria. This hypobaric hypoxia triggers a series of physiological responses, which, in most cases, help the individual tolerate and adapt to the low oxygen conditions. However, in other cases, maladaptive responses occur, that in turn cause one of three forms of acute altitude illness, acute mountain sickness (AMS), high-altitude cerebral oedema (HACE) and high-altitude pulmonary oedema (HAPE). These diseases can develop at any time from several hours to 5 days following ascent to a given elevation and can range in severity from mild with minimal effect on the planned travel itinerary to life-threatening illness.
The review is intended to provide detailed information about each of these entities. For each disease, we review the clinical features, epidemiology and the current understanding of their pathophysiology. We then review the primary pharmacological and nonpharmacological approaches to the management of each form of acute altitude illness and provide practical recommendations for both prevention and treatment. For the purposes of this review, we will consider high altitude as elevations ≥2500 m, although it should be recognised that the physiological responses to hypobaric hypoxia start at lower elevations, and thus some individuals who are highly susceptible to acute altitude illness may become sick at altitudes <2500 m.
AMS and HACE
Clinical aspects
AMS consists of nonspecific symptoms that occur at altitudes of ≥2500 m in unacclimatised individuals with a usual delay of 4–12 h after arrival at a new altitude. The symptoms are usually most pronounced after the first night spent at a new altitude and resolve spontaneously when appropriate measures are taken [1]. The leading symptom is headache, which is required for the diagnosis of AMS by the most frequently used scoring system [2]. However, this has been questioned by those who argue, based on anecdotal reports, that some individuals (probably ∼5% [3]) with symptoms clearly attributable to high altitude will be missed by having headache as a compulsory symptom. [4] Additional symptoms are loss of appetite or nausea, dizziness, fatigue or lassitude and insomnia. These are all nonspecific, and insomnia, in particular, is very prevalent in healthy individuals at high altitude. Progression of symptoms including nausea and headache not responding to first-line antiemetics and analgesics and increasing lassitude may point to progression of AMS to HACE [5]. The leading signs of HACE are truncal ataxia and clouded consciousness. Absence of preceding or concurrent headache does not exclude the diagnosis of HACE [6]. Without appropriate treatment, HACE usually leads to death, sometimes within 24 h of onset.
Clinical and routine laboratory examinations do not reveal abnormal findings in AMS. Usually, arterial oxygen saturation (SaO2) and tension (PaO2) are on average slightly lower compared to healthy controls at a given altitude, and the alveolar–arterial oxygen tension difference slightly greater (table 1). However, there is a large overlap of individual values [7] and statistically significant differences can only be obtained in studies of large groups. These blood gas changes might be due to a mild interstitial oedema not visible on chest radiographs [8], but suggested by a slight decrease of vital capacity and increase of closing volume [9, 10]; however, these are inconsistent findings [11]. Furthermore, a lower ventilatory drive in hypoxia might contribute to more severe hypoxaemia in AMS, at least in some individuals [12, 13]. Laboratory investigations of the brain in AMS show that all subjects exposed to altitudes of 4500 m had normal lumbar cerebrospinal fluid pressure and a small increase of brain volume assessed using MRI (<1%, ∼7–10 mL) after 16 h, independent of presence or absence of AMS [14]. The volume increase can partly be attributed to increased blood volume and partly to cell swelling [14, 15]. A recent study demonstrated swelling of white matter correlating with AMS scores after 22 h [16].
Arterial blood gas analysis at 4559 m
The clinical hallmarks of HACE are truncal ataxia and decreased consciousness, which can rapidly progress to coma without appropriate treatment, as discussed in the last section of this review. SaO2 and PaO2 are usually considerably lower than in AMS, even without signs of pulmonary oedema, which will eventually develop when HACE worsens [17]. Laboratory investigations show increased lumbar cerebrospinal fluid pressure [18]. Brain MRI performed at low altitude immediately after rescue by helicopter showed oedema in the corpus callosum [19]. Susceptibility-weighted MRI demonstrated a leak of the blood–brain barrier for erythrocytes as evidenced by hemosiderin deposition persisting over years in the corpus callosum and throughout the brain in more severe cases after HACE (figure 1) [20].

Susceptibility-weighted magnetic resonance imaging in a) axial and b) sagittal planes shows microhaemorrhages (arrows) in the corpus callosum of a 65-year-old female who had suffered from severe high-altitude cerebral oedema 7 weeks previously at 3450 m. Reproduced from [1] with permission from the publisher.
The differential diagnosis of AMS and HACE includes exhaustion, hypothermia, hyponatraemia, migraine, dehydration, infection, carbon monoxide poisoning, drug and alcohol intoxication, hypoglycaemia or severe hyperglycaemia, transient ischaemic attack or stroke and acute psychosis, possibly related to intake of corticosteroids [21].
Epidemiology
There is no systematic analysis of risk factors for HACE, due to the rarity of the disease. Since AMS may progress to HACE, risk factors for AMS are also relevant for HACE, as avoiding AMS will prevent HACE.
The major determinants of AMS are the altitude attained [22], individual susceptibility, rate of ascent and degree of preacclimatisation [13, 23, 24]. Therefore, prevalence of AMS depends very much on the study setting. Prevalence of AMS varies between 40% and 90%, depending on altitude and individual susceptibility in unacclimatised individuals ascending considerably more than 500 m·day−1 to altitudes of 4500–6000 m [23, 25], whereas prevalence with passive ascent to 3000–3500 m is ∼25–40% [26, 27]. HACE occurs rarely at altitudes <4000 m, and its prevalence between 4200 m and 5500 m is estimated to be 0.5–1% [17].
A low ventilatory drive in hypoxia [13, 27, 28] and history of migraine [13] are minor risk factors, while smoking and alcohol intake do not increase the risk of AMS. Aside from some limited evidence regarding lung disease [27] and obesity [27, 28], there is no evidence that the presence of underlying medical problems such as asthma, coronary artery disease or diabetes mellitus increase the risk of becoming ill following ascent. Susceptibility to AMS is not different between males and females, while children and adolescents are possibly less prone to AMS [26]. People aged >40–60 years tend to develop AMS less than younger adults [27]. Recent studies examining the hypothesis that exercise enhances AMS yielded controversial results [29–31].
Pathophysiology
We still lack a unified, coherent explanation for the development of AMS and HACE. Multiple factors have been implicated, as follows.
Hypoxaemia
Most studies measuring oxygenation in AMS find that SaO2 or PaO2 are lower in AMS versus non-AMS groups with a large interindividual overlap [32] (table 1). A lower ventilatory drive in hypoxia [7, 13], impaired gas exchange due to interstitial pulmonary oedema [10] and fluid retention [7] or increased metabolism [1] may contribute to slightly more pronounced hypoxaemia in AMS.
Consequences of more severe hypoxaemia in AMS might involve a greater increase in cerebral blood flow (CBF). CBF measured by transcranial Doppler in AMS yielded controversial results, possibly because of changes in the diameter of the middle cerebral artery that was assumed to remain unchanged [33]. However, changes in the diameter of the internal carotid artery did not correlate with high-altitude headache [34]. Thus it is not clear at present whether differences in CBF are present between those with and without AMS.
A further consequence of more severe hypoxaemia could be an increase in vascular permeability through a higher oxidative stress [35], low-grade inflammation [36] or increased hypoxia-inducible transcription factor-dependent expression of vascular endothelial growth factor (VEGF), which might be involved in the pathophysiology of HACE as suggested by animal experiments [37]. However, in AMS, lumbar punctures reveal an intact blood–brain barrier for large molecular weight proteins [38]. Furthermore, there are no significant associations of peripheral oedema [39], slight increase in brain volume [14], albuminuria [40] and leakage of fluorescent dye from retinal vessels [41] with AMS. Thus, the slight tissue swelling in the body in hypoxia is not different between those with and without AMS.
The brain
Headache is the primary symptom of AMS. Pain perception occurs only in the large blood vessels and meninges though sensory fibres of the trigeminal ganglia that project to the cortex. Connections of these afferent fibres to vegetative centres in the brainstem can explain accompanying symptoms such as nausea and vomiting [42]. Potential mechanisms of activation of these pain receptors in AMS are pressure or distortion through brain swelling or increased intracerebral pressure (ICP), vascular distension due to increased intravascular pressure or release of nociceptive chemicals/substances.
Most MRI studies in AMS found volume changes of <10 mL between 6 and 16 h of hypoxia with no correlation to AMS symptom scores [43]. These studies could not distinguish between oedema and changes in blood volume, but most of them found decreased water mobility compatible with intracellular swelling associated with AMS. A recent sequential MRI study over 22 h showed that increased inflow into the brain preceded formation of white matter oedema (13 mL), which correlated with a cumulative AMS score over 22 h [16]. Taken together, these studies indicate that AMS is accompanied by a mild intracellular oedema predominantly in the white matter that is unlikely to account directly for irritation of cerebral pain receptors.
Direct measurements indicate that ICP appears to be normal in subjects with AMS at rest [38, 44], but may increase with exercise or other factors that increase blood or intrathoracic pressure [45], due to decreased intracranial compliance. Disturbed autoregulation may occur in AMS [35, 46] and could enhance pressure transduction to the brain, although findings are controversial [47]. Repeated transient increases of ICP may sensitise trigeminal nociception [48] and decrease compliance of the CNS to a variable degree, since the capacity for spatial compensation varies considerably between individuals [49].
Venous outflow restriction due to anatomical variations of the sinus transversus leading to cerebral vein congestion was suggested to play a role in the pathophysiology of AMS [50]. However, the correlation between headache scores during trekking and retinal vein congestion at 5300 m reported in support of this hypothesis could not be confirmed in a more standardised setting at 4500 m [51]. In a recent article, those authors show that parenchymal swelling compresses small veins and hence also contributes to venous outflow restriction [16]. The concept of interindividual variability of venous outflow restriction contributing to AMS could explain the lack of consistent correlations between AMS and factors relevant for cerebral inflow, such as CBF, ventilator responses to hypoxia (HVR) or SaO2. It also fits with the observations that AMS worsens overnight or with bending over and improves after getting up and when CBF decreases with acclimatisation.
Thus, susceptibility to AMS appears to be determined by an interaction of physiological responses to hypoxia (ventilation, cerebral vasculature, autonomic nervous system and nociceptive thresholds) and anatomical factors such as the compensatory capacity for cerebrospinal fluid and the capacity of venous outflow. The mechanisms involved in the pathophysiology of AMS may also lead to HACE, which can be considered end-stage AMS. Longer duration of illness, development of worsened oxygenation with HAPE and greater hypoxic stress may account for progression. HACE is characterised by increased ICP, visually detectable oedema on MRI and a blood–brain barrier leak with hemosiderin deposition in the brain. VEGF is probably one of the major contributors to the severe vasogenic oedema leading to HACE, as suggested by animal experiments [37].
HAPE
Clinical aspects
HAPE occurs in healthy individuals at altitudes >2500–3000 m within 1–5 days after arrival [52]. It is rarely observed below these altitudes and after 1 week of acclimatisation. In many cases it is preceded by symptoms of AMS. Early symptoms include excessive exertional dyspnoea in relation to the patient's companions, mild cough, chest tightness and reduced exercise performance. As oedema progresses, cough and dyspnoea worsen and orthopnoea develops. Gurgling in the chest and pink frothy sputum indicate advanced cases.
Examination reveals cyanosis, tachypnoea, tachycardia, mildly elevated temperature and crackles upon auscultation. As SaO2 plummets, signs of hypoxic encephalopathy or, in some cases, HACE may develop. Arterial blood gas and SaO2 measurements in advanced HAPE at 4559 m demonstrate its severity: mean PaO2 registered in the mid-20 mmHg range versus 35–45 mmHg in healthy controls, and SaO2 measurements were <50% versus 70–85% (table 1) [53]. Chest radiographs and computed tomography scans in symptomatic HAPE (figure 2) show a patchy peripheral and nodular distribution of oedema [54]. Bronchoalveolar lavage (BAL) shows a protein-rich exudate and mild alveolar haemorrhage, which initially is noninflammatory, but may progress after several days to a more inflammatory picture, as discussed later [55–57]. Echocardiographic and pulmonary artery catheterisation studies at high altitude show marked pulmonary hypertension [58–60].

a) Chest radiograph of a 37-year-old male mountaineer with high-altitude pulmonary oedema (HAPE) showing a patchy to confluent distribution of oedema, predominantly on the right side; b) computed axial tomography scan of 27-year-old mountaineer with recurrent HAPE showing patchy bilateral nodular distribution of oedema. Reproduced from [61] with permission from the publisher.
Epidemiology
Two populations are affected by HAPE. The first involves well-acclimatised alpine residents returning from low altitudes (re-entry HAPE) and the second involves rapid ascent of unacclimatised lowlanders. Altitude, ascent rate and individual susceptibility are the major determinants of HAPE. Its prevalence ranges from <0.2% in a general mountaineering population when climbing in ≥3 days to altitudes of 4000–5000 m, but as high as 7% with a single-day ascent. A similar increase in HAPE incidence of 2.5% versus 15.5% occurs when an altitude of 5500 m is reached by trekking over 4–6 days as opposed to airlift. In those with a history of radiographically documented HAPE, the likelihood of developing HAPE is 60% with a 1–2-day ascent to the same altitude [52, 53].
Pathophysiology
The pathophysiology of HAPE has been reviewed extensively elsewhere [61]. Since its recognition in the 1960s, a number of pathophysiological mechanisms have been proposed. It was clear early on that pulmonary hypertension and HAPE were inextricably linked, suggesting a primary haemodynamic basis. However, it is clear that some individuals with strong hypoxic pulmonary vasoconstriction (HPV) do not develop HAPE [62]. Thus, other factors may be necessary contributors to HAPE susceptibility. Studies suggestive of an inflammatory pathogenesis, reduced or defective active sodium and water reabsorption by the hypoxic alveolar epithelium and unevenness of regional HPV have been advanced as possibilities for how HAPE may develop.
Haemodynamics
Right heart catheterisation at high altitude revealed mean (range) pulmonary artery pressures of 60 (35–115) mmHg, but normal pulmonary artery wedge pressures [58–60]. These pioneering studies put to rest the idea that HAPE was acute left heart failure at high altitude. Excessive pulmonary artery pressures precede the development of HAPE and are not a consequence of the disease [63]. The critical role of high pulmonary artery pressure is further confirmed by the fact that descent, oxygen or drugs that lower pulmonary artery pressure are effective for prevention and treatment as discussed below.
Individuals susceptible to HAPE have many physiological characteristics that place them at greater risk. The most important is a strong HPV response, as well as higher pressures with even normoxic exercise [64, 65]. The exaggerated rise in pulmonary artery pressure in susceptible individuals developing HAPE is accompanied by increased microvascular pressure >20 mmHg, a threshold pressure for formation of alveolar oedema [59, 66].
The basis for high hypoxic pulmonary artery pressures in HAPE-susceptible subjects is multifactorial. They have lower HVR [12, 67] and stronger sympathetic tone [68]. HVR is set largely by the peripheral chemoreceptors, which results in a lower alveolar oxygen tension and higher carbon dioxide tension at the same altitude as HAPE-resistant subjects, and thus leads to a stronger stimulus for HPV [69]. Increased peripheral hypoxic chemoreceptor sensitivity also appears to reduce the strength of HPV, independent of differences in alveolar ventilation [70]. HAPE-susceptible individuals have slightly lower lung volumes and reduced diffusion capacity [12, 71, 72].
In addition, there are differences in the vasculature. Nitric oxide (NO) and endothelin-1 are important pulmonary endothelial-derived vasodilator and vasoconstrictor mediators. Endothelin production is greater in HAPE susceptibility [73], while lung NO production is reduced [56, 74, 75]. Systemic vascular endothelial NO generation is reduced more in hypoxic HAPE-susceptible subjects compared to controls [76]. This is probably also the case for the pulmonary circulation, as the above-cited measurements of exhaled NO and metabolites of NO in lavage fluid suggest.
The question arises how hypoxic constriction of pulmonary vessels leads to oedema. Three mechanisms have been suggested [53]: 1) transarteriolar leakage with oedema occurring upstream of the microvasculature at small weaker arteriolar right-angle branches off larger pulmonary arteries; 2) heterogeneous regional arterial hypoxic vasoconstriction; and 3) hypoxic venoconstriction. If arterial vasoconstriction in hypoxia is inhomogeneous, HAPE could be the consequence of higher flows in those areas with lesser vasoconstriction, leading to increased microvascular pressure. MRI of lung blood flow demonstrates that HPV is uneven at rest in susceptible individuals [77, 78], but not in those with proven HAPE resistance. This unevenness in regional HPV may help to explain the characteristic patchy and nodular appearance of oedema in HAPE.
The reason(s) why the pulmonary vasculature leaks under high pressure are not completely resolved. Traditionally, pulmonary oedema has been categorised as either noncardiogenic (increased permeability with exudative characteristics: high protein concentrations and markers of inflammation in the setting of normal or only modestly elevated intravascular pressures) or cardiogenic (elevated hydrostatic pressures leading to a noninflammatory protein-poor transudative leak). As described above, BAL fluid in nascent HAPE [56] reveals characteristics of a hydrostatic, but noncardiogenic noninflammatory oedema suggesting pressure-induced alterations to the normal permeability of the alveolar–capillary barrier or frank traumatic injury; the latter has been termed capillary stress failure [79].
If hydrostatic forces persist, then gene upregulation and transcription of collagen and other extracellular matrix proteins are initiated to strengthen the alveolar capillary barrier [80] and ultimately reduce stress failure and leak. These observations offer an explanation for the rapid recovery from HAPE and the protection from recurrence when re-ascending only several days after recovery from HAPE.
Inflammation
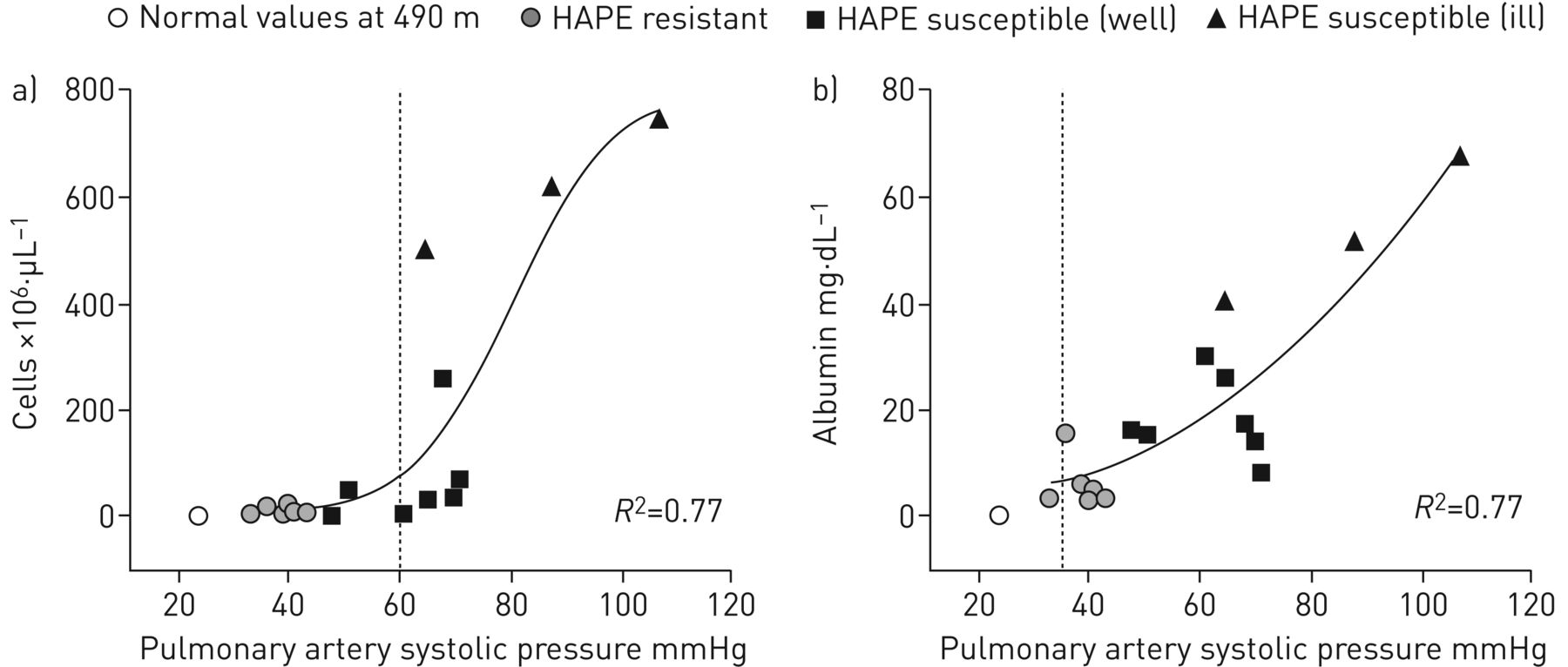
The first measurements of alveolar lavage fluid in mountaineers [55, 57] were obtained in those with established HAPE. In addition to the high protein concentrations, some, but not all cases had significant neutrophilia and elevations of pro-inflammatory cytokines and neutrophil chemotactic factors. These observations strongly suggested that inflammation might be a causal factor in HAPE, leading to a greater permeability of the lung microvasculature. However, not all cases of HAPE had evidence of inflammation [55], indicating that in humans inflammation is possibly a secondary response to alveolar–capillary barrier disruption or oedema. BAL in HAPE-susceptible climbers showed mild alveolar haemorrhage and increased serum-derived protein concentrations within a day of ascent to 4559 m from low altitude both in subjects ill with HAPE at the time of bronchoscopy and in those who developed HAPE in the next 24 h [56]. There is a strong correlation between the magnitude of pulmonary hypertension assessed by echocardiography and the degree of haemorrhage and protein elevation in the alveolar space (figure 3). In contrast, there were no increases in alveolar macrophages, neutrophils and pro-inflammatory mediators at high altitude early in the course of HAPE.
{kind=link}
{kind=link}
{kind=link}
Individual bronchoalveolar lavage a) red blood cell counts and b) albumin concentrations plotted against pulmonary artery systolic pressures at high altitude (4559 m). Reproduced from [56] with permission from the publisher.
It is not clear what initiates the secondary inflammation. It may be that sustained and increasing high pressures of sufficient duration in untreated HAPE can trigger inflammation [81], or it represents part of the healing process of a markedly disrupted alveolar–capillary barrier that occurs in the most severe cases of HAPE, especially with alveolar haemorrhage, since haem and other breakdown products of red cell haemoglobin are chemotactic for neutrophils [82]. Despite overwhelming evidence against a primary inflammatory alteration of the alveolar–capillary barrier in HAPE, it is nevertheless conceivable that any concurrent process altering the permeability of the alveolar–capillary barrier will lower the pressure required for formation of oedema. Indeed, increased fluid accumulation during hypoxic exposure after priming by endotoxin or virus in animals [83, 84] and the association of preceding respiratory viral infections with HAPE in children [85, 86] support this concept. Thus, upper respiratory tract infections shortly before a sojourn in the mountains and vigorous exercise at altitudes between 2000 and 3000 m may explain in some cases why HAPE can develop at a modestly low altitude [87].
Alveolar fluid clearance
Disruption of the alveolar capillary barrier and leak are the proximal causes of HAPE, but studies have highlighted the possibility that alveolar fluid clearance mechanisms dependent upon active alveolar epithelial sodium and water reabsorption by type I and II pneumocytes may contribute to the pathophysiology of HAPE. Active fluid transport from the alveolar space into the lung interstitium and clearance via lymphatics is important in normal lung fluid balance. Hypoxia decreases transepithelial sodium transport by reducing expression and activity of the epithelial sodium channel (ENaC) and sodium (Na+)/potassium (K+) ATPase proteins [88], possibly by an impairment of β2-adrenergic receptor signalling [89]. In vivo hypoxia depresses alveolar fluid clearance in hypoxic animals [90, 91]. Mice partially deficient in ENaC develop greater accumulation of lung water in hypoxia [92].
Another approach to study the relevance of alveolar transepithelial fluid reabsorption in HAPE has involved the use of β2-receptor agonists and glucocorticoids, both of which upregulate ENaC and Na+/K+ ATPase [93]. Two field studies reported successful prevention of HAPE in HAPE-susceptible climbers with inhalation of salmeterol, a long-acting β2-agonist [94], and oral dexamethasone [95] begun 1 day before ascent. Owing to multiple actions of β2-adrenergic agonists, such as inhibition of HPV, increased HVR and ventilation, tightening of cell-to-cell contacts and upregulation of NO production [96], the contribution of enhanced alveolar fluid clearance remains uncertain. Indeed, the protective effect of dexamethasone from a putative enhancement of alveolar fluid reabsorption could not be correlated with indirect measures of enhanced active sodium reabsorption, but rather to a surprising reduction of pulmonary artery pressure [95].
Prevention of acute altitude illness
Depending on the circumstances of the planned ascent, prevention of acute altitude illness is based on a combination of various nonpharmacological and pharmacological approaches.
Predicting who will get sick
For the purpose of preventing acute altitude illness, it would be helpful to predict who is at risk of developing one of the three acute altitude illnesses. For an individual who has been to high altitude before, past performance is a good, but not perfect, predictor of future performance upon return to a similar altitude at a similar rate. For example, Bärtsch et al. [97] have shown that ∼60% of HAPE-susceptible individuals develop HAPE on subsequent trips to high altitude. However, there remains no easy way to predict how the altitude-naïve traveller will fare following ascent. Canoui-Poitrine et al. [98] have developed and validated a model for identifying individuals at risk for severe altitude illness (severe AMS, HACE or HAPE), but the model requires an individual to undergo an exercise test while breathing a hypoxic gas mixture and, as a result, is not amenable to widespread, easy application. Furthermore, the value of such testing, which at present has not been validated by a prospective trial, was questioned in a recent pro [99] and con [100] debate.
Nonpharmacological measures
Slow ascent
Because overly rapid ascent remains the primary risk factor for developing acute altitude illness, undertaking a slow ascent to the target elevation remains the best method for altitude illness prevention. With ascents >2500–3000 m, individuals should not increase their sleeping elevation by >300–600 m per night and should take a rest day every 3–4 days, in which they sleep at the same elevation for at least one additional night. This recommendation, the specifics of which vary slightly depending on the resource cited [52, 101, 102], derives largely from observational data [103, 104] and has only been examined in a prospective, randomised manner in a single study [24]. Because local terrain and logistical factors often prevent adherence to these specific altitude limits on a day-by-day basis, individuals should instead focus on the ascent rate averaged over the entire trip [105]. Due to the significant interindividual variability in susceptibility to altitude illness, some individuals can tolerate faster ascent profiles than those prescribed above. People who travel repeatedly to high altitude learn more about their personal tolerances and can adjust their ascent rates accordingly and perhaps ascend at faster than recommended rates, while the individual with no prior experience in this environment should adhere to those limits specified above.
Preacclimatisation
Spending time at moderate altitude before ascending to the target elevation, described as “staged ascent”, augments the beneficial physiological responses seen following ascent and decreases the incidence of acute altitude illness [106]. Studies have reported conflicting results regarding intermittent normobaric or hypobaric hypoxic exposures, with some studies showing benefit [107] and others not demonstrating a clear effect [108, 109]. One of the challenges in interpreting these discrepant results is that the hypoxic exposure protocols vary significantly between studies with regard to the magnitude and duration of the hypoxic exposures. While these preacclimatisation strategies may carry benefit and probably pose little risk, implementation may be difficult for many travellers and the optimal approach remains unclear. Infrequent and/or short exposures to hypoxia are unlikely to reduce the incidence of AMS [109, 110], while longer and/or more frequent exposures are necessary to reduce the risk of altitude illness [107, 111]. Such exposures should be performed as close in time to the planned high-altitude travel as possible. One particular preacclimatisation strategy that has received recent attention is sleeping at low elevation in an enclosed, oxygen-depleted space (e.g. a tent enclosing the bed) on a nightly basis. While anecdotal reports suggest that the use of such systems is increasing among climbers and other individuals travelling to high altitudes, the utility of this approach has not been examined extensively. Dehnert et al. [112] conducted a randomised trial which demonstrated reduced symptoms and incidence of AMS with 14 consecutive nights of sleep in normobaric hypoxia, but reported technical difficulties maintaining the desired level of hypoxia on a consistent basis.
Other measures
Avoiding excessive alcohol consumption and opiate pain medications are other common-sense measures for preventing acute altitude illness, but which have not been studied in a systematic manner. Avoiding overexertion following ascent is another commonly recommended measure, although studies have reported varying results about the relationship between exertion and risk of AMS [29, 30, 113]. Abrupt cessation of caffeine intake in chronic users of caffeinated beverages may provoke withdrawal symptoms that mimic those of AMS [114]. The risk of dehydration is increased at high altitude due to the lower humidity in this environment and decrease in plasma volume following ascent, but dehydration has not been shown to increase the risk of AMS [115, 116]. Fluid intake should be driven by thirst and sufficient to prevent dehydration and avoid continuously low and highly concentrated urine output, but systematic attempts to overhydrate should be avoided.
Pharmacological measures
A variety of medications can be used for the prevention of acute altitude illness (table 2). However, pharmacological prophylaxis is not necessary in all high-altitude travellers, and instead should be initiated based on an assessment of the risk of acute altitude illness associated with a planned trip that accounts for the individual's prior performance at high altitude, if known, and features of the planned ascent, such as the altitude reached on the first day, the number of planned rest days and the anticipated increase in sleeping elevation once >2500–3000 m (table 3) [52, 102]. Pharmacological prophylaxis should be strongly considered for moderate–high-risk ascent profiles, but is not necessary in low-risk situations.
Medications for the prevention and treatment of acute altitude illness
Risk assessment for acute altitude illness
Pharmacological prophylaxis is largely directed toward the prevention of acute mountain sickness, which, as noted earlier, is by far the most common problem that affects people following ascent. At the same time, because HACE and HAPE are reflective of impaired acclimatisation, medications that prevent AMS theoretically reduce the risk of these entities as well, but this concept has not been studied in a systematic manner.
Acetazolamide remains the mainstay of AMS prophylaxis. While there has been debate about the appropriate dosage for prophylaxis [117], the bulk of the evidence suggests 125 mg twice daily is sufficient for most climbs [102, 118], but may be inadequate for overly rapid ascents and/or ascents to very high final altitudes [119]. The medication is typically started the night before the planned ascent and continued until descent is initiated or until the individual has been at the target elevation for 2–3 days. Dexamethasone is a well-studied alternative [120, 121] for those who are intolerant of or have a contraindication to acetazolamide. Given the risk of adrenal suppression with long-term use of high-dose systemic corticosteroids, the medication should not be used for more than seven consecutive days and, if longer use is necessary, should be tapered off rather than stopped abruptly [21, 102].
Recent studies have suggested that ibuprofen at a dose of 1800 mg·day−1 may be effective at preventing AMS [122], but studies have yet to establish superiority over acetazolamide or dexamethasone. In addition, safety with prolonged use at high altitude and, in particular the risk of gastrointestinal bleeding, remain unclear. Other recent studies have suggested that the inhaled steroid budesonide may have a role in AMS prophylaxis [123, 124]. These studies raise important questions about lung involvement in the underlying pathophysiology of AMS [125], but have yet to be replicated and do not provide sufficient rationale for using this expensive medication in place of acetazolamide or dexamethasone.
As noted in table 2, medications are available specifically for HAPE prophylaxis, but these are generally reserved for those with a prior history of HAPE. Based on a small, randomised trial [126] and extensive clinical experience, the pulmonary vasodilator nifedipine is the primary medication for this purpose. Other data demonstrate that the phosphodiesterase inhibitor tadalafil, dexamethasone [95] and the long-acting β2-agonist salmeterol [94] are also effective at preventing HAPE in known susceptible individuals, although the latter agent is less effective than pulmonary vasodilators and may be considered only for use as an adjunct to other agents and not as the sole prophylactic option [102]. The dose is also substantially higher than that used for asthma treatment and can lead to tachycardia and tremulousness.
Individuals who are not familiar with the prophylactic medications should consider taking one or two doses prior to their planned travel to assess their tolerance of the medications and side-effect profile.
Treatment
While descent to lower elevation is the best treatment for all forms of acute altitude illness, the optimal approach varies based on the type and severity of illness and other factors.
AMS
The majority of patients who become ill at high altitude have AMS, which can typically be treated by stopping ascent and using nonsteroidal anti-inflammatory drugs acetaminophen or aspirin for headache and, in some cases, antiemetics for nausea. The utility of these drugs for treating the full spectrum of AMS symptoms has not been specifically examined, but they have been shown to be effective at treating high-altitude headache [127, 128]. Rehydration does not treat AMS per se, but does address any dehydration, the symptoms of which can mimic those of AMS. Acetazolamide [129] and dexamethasone [121, 130] can be added for those individuals with more severe AMS symptoms or those who fail to respond to conservative measures. Individuals who remain ill despite several days of these conservative measures should descend 500–1000 m or until symptoms resolve.
HACE
The onset of neurological symptoms suggestive of HACE is an indication for immediate descent. If not feasible due to weather, terrain or other logistical factors, ill individuals should be placed on supplemental oxygen via either an oxygen tank or oxygen concentrator or placed in a portable hyperbaric chamber [131]. Dexamethasone should be administered by the oral, intramuscular or intravenous route at a dose of 8 mg initially followed by 4 mg every 6 h until the person has descended or symptoms have fully resolved. [102] Diuretics are not part of the standard approach for HACE, nor is there any evidence or rationale for acetazolamide.
HAPE
The optimal approach to HAPE varies based on whether the individual is able to access care in a well-resourced setting [102, 132]. Fully conscious patients who can access health facilities in mountain resorts, for example, often do not need to descend, and instead can be managed with supplemental oxygen alone and close observation at either the health facility or their lodge. However, in more remote and/or under-resourced settings, HAPE patients should descend to lower elevation or, if not feasible, be treated with supplemental oxygen or a portable hyperbaric chamber as well as a pulmonary vasodilator. Based on a small, uncontrolled study [133], sustained-release nifedipine is the preferred agent for this purpose, while anecdotal reports suggest that phosphodiesterase-5 inhibitors may be useful as well (table 2). Cases series describe concurrent use of pulmonary vasodilators, acetazolamide and inhaled β2-agonists in HAPE treatment [134, 135], but there are no data to support this approach. While very early reports described their use in HAPE [136], diuretics are not part of standard protocols because many patients are volume depleted at the time of their illness, and diuretic administration increases the risk of hypotension. Use of continuous positive airway pressure without supplemental oxygen has also been described in several reports [87, 137], but has not been studied in a systematic manner.
Re-ascent following resolution of acute altitude illness
Individuals affected by AMS can resume their ascent once symptoms have resolved with careful attention to the rate of ascent and consideration of pharmacological prophylaxis. Although reports document successful ascents of Mount Everest following episodes of HAPE [138, 139], the safety of continuing ascent after resolution of HAPE or HACE remains controversial. If ascent is pursued in such circumstances, the individual should be symptom-free and off any medications for at least several days before initiating further ascent and should strongly consider pharmacological prophylaxis for their ascent, including a pulmonary vasodilator for individuals who had HAPE and dexamethasone for individuals who had HACE.
Footnotes
Previous articles in this series: No. 1: Adir Y, Bove AA. Can asthmatic subjects dive? Eur Respir Rev 2016; 25: 214–220. No. 2: Szpilman D, Orlowski JP. Sports related to drowning. Eur Respir Rev 2016; 25: 348–359. No. 3: van Ooij PJAM, Sterk PJ, van Hulst RA. Oxygen, the lung and the diver: friends and foes? Eur Respir Rev 2016; 25: 496–505. No. 4: Mijacika T, Dujic Z. Sport-related lung injury during breath-hold diving. Eur Respir Rev 2016; 25: 506–512.
Conflict of interests: None declared.
Provenance: Submitted article, peer reviewed.
- Received September 13, 2016.
- Accepted October 23, 2016.
- Copyright ©ERS 2017.
ERR articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










