





Abstract
Pulmonary hypertension (PH) associated with left heart disease (PH-LHD) is the most common type of PH, but its natural history is not well understood. PH-LHD is diagnosed by right heart catheterisation with a mean pulmonary arterial pressure ≥25 mmHg and a pulmonary capillary wedge pressure >15 mmHg. The primary causes of PH-LHD are left ventricular dysfunction of systolic and diastolic origin, and valvular disease. Prognosis is poor and survival rates are low. Limited progress has been made towards specific therapies for PH-LHD, and management focuses on addressing the underlying cause of the disease with supportive therapies, surgery and pharmacological treatments. Clinical trials of therapies for pulmonary arterial hypertension in patients with PH-LHD have thus far been limited and have provided disappointing or conflicting results. Robust, long-term clinical studies in appropriate target populations have the potential to improve the outlook for patients with PH-LHD. Herein, we discuss the knowledge gaps in our understanding of PH-LHD, and describe the current unmet needs and challenges that are faced by clinicians when identifying and managing patients with this disease.
Abstract
Pulmonary hypertension due to left heart disease is associated with multiple unmet medical needs http://ow.ly/TFET8
Case study part 1: initial assessment
A 63-year-old male with hypertension, diabetes mellitus, dyslipidaemia, coronary artery disease and obstructive sleep apnoea presented to the clinic with worsening dyspnoea upon exertion (World Health Organization (WHO) functional class IIIb). Echocardiography revealed a normal-sized left ventricle, left ventricular ejection fraction (LVEF) of 35–40%, delayed left ventricular diastolic relaxation, normal left atrium, right ventricular dilation with moderately depressed systolic function, mild right atrial enlargement, flattened interventricular septum, dilated inferior vena cava, and a pulmonary artery systolic pressure of 85 mmHg. Computed tomography pulmonary angiography revealed no pulmonary embolus or parenchymal lung disease. The patient failed to reach the target heart rate in a treadmill exercise test, but there was no evidence of myocardial ischaemia. Pulmonary function testing and right heart catheterisation (RHC) were performed (table 1).
Case study: results from pulmonary function testing and right heart catheterisation
Question 1: Based on these data, what would be your initial assessment of this patient?
This real-life patient case study highlights some of the many factors that can make diagnosing pulmonary hypertension associated with left heart disease (PH-LHD) challenging. Based on the information presented, would you diagnose this patient with PH-LHD?
In this review, we will discuss the challenges that are associated with diagnosing and managing PH-LHD, before revisiting the case study at the end of this article. The topics that will be presented here might change your evaluation of this patient's diagnosis.
Introduction
PH-LHD is a common form of pulmonary hypertension (PH) that is associated with a poor prognosis and high rate of morbidity and mortality [1, 2]. However, there is still a lack of understanding about its pathophysiology, epidemiology and treatment [1–14]; as such, there are numerous unmet needs for this condition (table 2). Multiple disorders are responsible for PH-LHD, including: 1) left ventricular systolic dysfunction (heart failure with reduced ejection fraction (HFrEF)); 2) left ventricular diastolic dysfunction (heart failure with preserved ejection fraction (HFpEF)); 3) left-sided valvular disease; and 4) congenital/acquired left heart inflow/outflow tract obstruction and congenital cardiomyopathies [2, 5]. In many cases of PH-LHD, there is a degree of overlap in these aetiologies [5].
Unmet needs for pulmonary hypertension associated with left heart disease (PH-LHD)
PH-LHD is characterised by backwards transmission of filling pressures due to impaired left ventricular diastolic function. A loss of left atrial compliance, exercise-induced mitral regurgitation and diastolic dysfunction, combined with increased pulsatile load caused by the increased pulmonary artery wedge pressure (PAWP), can lead to and/or exacerbate the PH [4]. Increases in mean pulmonary arterial pressure (PAP) in certain individuals may trigger development of PH. In some patients, additional factors, such as endothelial dysfunction (resulting in decreased nitric oxide availability and increased expression of endothelin) [15], an altered response to brain natriuretic peptide (BNP) (impacting vascular tone) [16], and vasoconstriction and/or vascular remodelling [3], can lead to further increases in vascular resistance and, hence, raised PAP [4]. As such, extensive vascular remodelling can occur in PH-LHD, resulting in decreased vascular compliance; right ventricle overload and right ventricular failure can then ensue, eventually leading to death [4]. It is important to remember that the most common cause of right ventricular dysfunction is still chronic heart failure [17].
There is no consensus regarding the true prevalence of PH-LHD in heart failure; it is estimated that up to 80% of patients with heart failure develop PH, depending on the methodology used to provide the estimates [18–23]. PH is common in patients with HFpEF and can be severe; ∼50% of patients with heart failure will have HFpEF [18]. Depending on the method of detection, PH has been shown to occur in ≥60% of patients with systolic dysfunction [6, 24]. In patients undergoing heart transplantation, PH has been reported as the cause of perioperative death in 44% of cases. Furthermore, the presence of PH could be considered a contraindication for carrying out transplantation [25].
In this review, we discuss the knowledge gaps in our understanding of PH-LHD and challenges faced by clinicians in the diagnosis and management of patients with PH-LHD. This review also provides an overview of how PH-LHD is currently managed in patients, and reflects on a number of potential strategies that might improve patient outcomes.
Classification and diagnosis of PH-LHD
Differential diagnosis of PH-LHD from other forms of PH can be highly challenging due to misunderstandings about the determinants of the disease, heterogeneity in the definitions and terminology, and variations in the different haemodynamic presentations. PH-LHD belongs to group 2 of the World Symposium on Pulmonary Hypertension PH clinical classification [26]. This group is defined as post-capillary PH, characterised by a mean PAP of ≥25 mmHg and PAWP of >15 mmHg (fig. 1) [26, 27]. In contrast, the haemodynamic definition of pre-capillary PH differs from post-capillary PH in that PAWP is ≤15 mmHg [27]. Pulmonary arterial hypertension (PAH) (group 1), PH due to pulmonary disease (group 3), chronic thromboembolic PH (group 4), and PH with unclear and/or multifactorial mechanisms (group 5) are all types of pre-capillary PH [27]. Thus, group 2 PH is the only current classification that is characterised purely by post-capillary PH (some of the entities in group 5 can involve both post- and pre-capillary mechanisms).
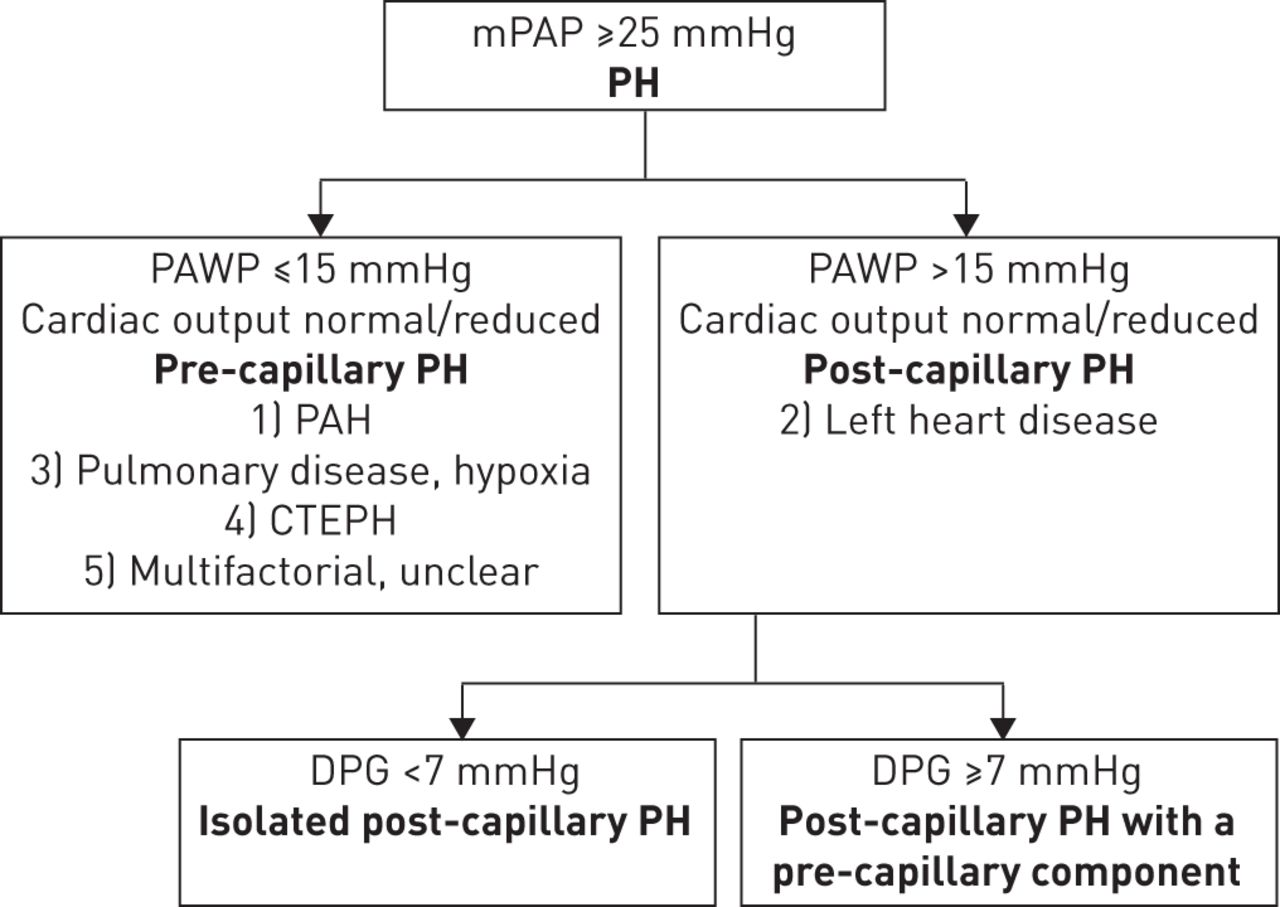
{kind=link}
Definition of pulmonary hypertension (PH) associated with left heart disease. mPAP: mean pulmonary arterial pressure; PAWP: pulmonary artery wedge pressure; PAH: pulmonary arterial hypertension; CTEPH: chronic thromboembolic pulmonary hypertension; DPG: diastolic pressure gradient. Information from [4, 26, 27].
Challenges in diagnosis
A major challenge associated with diagnosing PH-LHD is the variation in terminology. PH is a known result of left-sided heart failure, regardless of the cause [18]. When differentiating HFpEF from HFrEF it is important to consider that although patients in both subgroups will present with signs and symptoms of heart failure, HFrEF is associated with a marked reduction in LVEF, whereas HFpEF is associated with a normal LVEF [18]. Management strategies and outcomes for patients with HFpEF and HFrEF may differ; however, differentiating between these two states can be challenging [28].
Differentiating between pre- and post-capillary PH, especially in patients with HFpEF, can also be diagnostically challenging. Misdiagnosis of PH-LHD as PAH could result in patients receiving inappropriate, or even detrimental, therapies [4]. In addition, a previously elevated but now normalised PAWP (with persistence of pulmonary vascular disease) can be seen in patients who have received diuretics and/or undergone the overnight fast associated with catheterisation. This is often the case in patients with HFpEF [4], leading to confusion between group 2 and group 1 PH patients at the time of catheterisation.
Variations in how to describe the different haemodynamic components is another potential cause of confusion in PH-LHD. Previous definitions categorised patients as having “passive” or “reactive” (also termed “out of proportion”) disease [4, 29]. An updated classification for post-capillary haemodynamics has recently been proposed [4]. This method takes into account the diastolic pressure gradient (DPG) (defined as the difference between diastolic PAP and mean PAWP), and defines patients with a low DPG (<7 mmHg) and PAWP >15 mmHg as having isolated post-capillary PH, but patients with DPG ≥7 mmHg and PAWP >15 mmHg as having post-capillary PH with a pre-capillary component (fig. 1) [4].
What is the advantage of this new definition?
Defining PH-LHD based on DPG has several advantages in classifying patients, including the fact that DPG is low in healthy individuals and is relatively simple to calculate. Relying on measurement of the DPG to distinguish between post-capillary PH with a pre-capillary component enables assessment of a patient's status while avoiding assumptions about the haemodynamic components of vasoconstriction and remodelling [23]. An association between elevated DPG and vascular remodelling, which is associated with significant pulmonary vascular disease and increased risk of mortality, has been demonstrated by a single study [30]. However, the prognostic benefit of this measurement might be limited, as factors other than vascular remodelling can lead to increases in DPG [31]. In a large study of patients with heart failure undergoing heart transplantation, DPG did not predict post-transplant survival [31]. In the 2015 guidelines for the diagnosis and treatment of PH [27], the joint task force of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS) recommends that a combination of pulmonary vascular resistance (PVR) and DPG should be used to define the different types of PH-LHD: isolated post-capillary pulmonary hypertension (DPG <7 mmHg and/or PVR ≤3 Wood units) and combined post-capillary and pre-capillary pulmonary hypertension (DPG ≥7 mmHg and/or PVR >3 Wood units). Furthermore, these guidelines state that patients with PH-LHD who have a high DPG and/or PVR should be referred to an expert PH centre for a complete diagnostic work-up and decisions about treatment [27].
Diagnostic methods
Several invasive and noninvasive methods can be used for diagnosis of suspected PH-LHD [2]. An understanding of patient comorbidities and disease risk factors can greatly assist with the differential diagnosis from other conditions and types of PH. Medical history review can help determine the clinical course and evaluate risk factors such as orthopnoea and paroxysmal nocturnal dyspnoea [2]. Physical examination can detect pulmonary oedema and pleural effusions [2], as well as signs of cardiac enlargement, left-sided S3 or S4 and murmurs consistent with valvular disease [6]. If PH is suspected, echocardiography can evaluate the patient's ventricular and valve function [6]. Laboratory tests can rule out anaemia and renal, thyroid or hepatic dysfunction [6]. The level of BNP and N-terminal pro-brain natriuretic peptide (NT-proBNP) can also be measured [6]; in patients with acute HFpEF a high level of NT-proBNP is a risk factor for poor short-term prognosis [32].
Cardiac catheterisation can be used to distinguish pre- and post-capillary PH [23, 33, 34]. As well as assessing pulmonary haemodynamics, catheterisation can be used to identify coronary artery disease and valvular disease and to measure left-sided filling pressure, if necessary by direct measurement of left ventricular pressure [4, 6]. One of the pitfalls of relying on RHC for differentiating PAH from PH-LHD is that most patients will be in a fasted and diuresed state, which can lead to underestimation of left heart filling pressure [22]. Administration of ∼500 mL saline to patients undergoing RHC has been shown to increase PAWP, which could help differentiate patients with PAH from those with occult left ventricular dysfunction [35]. However, as yet, this technique has not been validated, although it has recently been recommended [36].
Vasoreactivity testing can be used to assess PH reversibility in cardiac transplant candidates [6]. This can be carried out at the same time as RHC, and involves the administration of parenteral nitroprusside, nitroglycerin or inhaled nitric oxide, with close titration, to achieve a positive vasodilator response (reduction in PVR to <2.5 Wood units) [37]. However, vasoreactivity testing in nontransplant candidates with PH-LHD has not been investigated and is currently not recommended, as there is currently no internationally accepted protocol for this procedure [4, 33].
Prognosis
PH-LHD is associated with a poor prognosis, and high risk of morbidity and mortality [3, 29, 38–40]. The 12-month mortality rates for PH-LHD have been estimated to be as high as 32%, with predictors of mortality including older age, male sex, right ventricular dysfunction, renal disease and lower functional class [41]. Among patients with advanced heart failure, reduced right ventricular ejection fraction (RVEF) (using a value of <35% as a surrogate for right ventricular systolic dysfunction) is associated with a high risk of death or the need for urgent transplantation, whereas patients with preserved RVEF have a prognosis that is very similar to patients with normal PAP [39]. Furthermore, in a large cohort of patients with chronic HFrEF (LVEF ≤40%), those with pre-capillary PH, as determined by PVR ≥3 Wood units, mean PAP ≥25 mmHg and PAWP ≥15 mmHg, were found to have a greater risk of death than patients with PH-LHD (PVR <3 Wood units, mean PAP ≥25 mmHg and PAWP ≥15 mmHg) (hazard ratio 1.55, 95% CI 1.11–2.20; p<0.001), suggesting that PH-LHD in HFrEF carries a greater risk of death than when other types of PH are present [42]. Among patients with PH-LHD who have acute decompensated heart failure, 6-month survival rates are lower in patients with post-capillary PH with a pre-capillary component than those with isolated post-capillary PH (48.3% versus 21.8% mortality, respectively) [38].
Prognosis in patients with heart failure can be determined by measuring RVEF [43–46]. In moderate congestive heart failure, RVEF has been shown to be an independent predictor of survival, with 1-year survival rates for RVEF <25%, ≥25% to <35% and ≥35% of 80%, 90% and 95%, respectively [43]. Moreover, increased PAP (mean PAP >20 mmHg) coupled with low RVEF (<35%) is associated with lower patient survival than normal PAP plus preserved/low RVEF or high PAP/preserved RVEF [39]. In patients with PH-LHD due to HFpEF, right ventricular systolic dysfunction is associated with higher mortality than patients with pulmonary artery systolic pressure >47 mmHg, even after adjustment for age and pulmonary artery systolic pressure [29].
Management of PH-LHD
Although a treatment algorithm has been developed for group 1 PH (PAH) in the ESC/ERS guidelines, it does not apply to patients with PH-LHD [27]. Prior to the assessment of patients for PH-LHD, the ESC/ERS guidelines recommend that treatment of underlying conditions should be optimised and any other causes of PH should be ruled out [27]. General supportive therapies for PAH include supplemental oxygen, diuretics, oral anticoagulants and digoxin, and specific drug therapy includes calcium channel blockers, prostanoids, endothelin receptor agonists, soluble guanylyl cyclase stimulators and phosphodiesterase type-5 inhibitors [27]. There is currently no similar approved specific therapy for PH-LHD; furthermore, clinical trial data for these patients are lacking [2–4]. Managing the underlying cause of PH is an important first step in treating PH-LHD [4]. For example, in the case of left heart disease due to valve disease, surgical repair or replacement of mitral or aortic valves can improve patient outcomes [2, 3, 47]. Reducing left heart filling pressures can also be achieved using established treatments for heart failure [2]. Diuretics are the mainstay of medical treatment for fluid control and relief of congestion, whereas angiotensin-converting enzyme inhibitors and β-blockers are indicated to improve outcomes in heart failure associated with HFrEF [23].
Several clinical trials have investigated the use of PAH therapies (e.g. prostanoids, endothelin receptor antagonists and phosphodiesterase type 5 inhibitors) in patients with suspected PH-LHD [48–60]; however, there is no clinical evidence to support the use of PAH therapies in the clinical management of patients with PH-LHD [27]. The results of studies that have investigated PAH-specific therapies in PH-LHD (many of which are summarised in table 3) have generally been negative and some could even be regarded as harmful. Furthermore, some of the trials that have been carried out have been prospective, randomised, placebo-controlled studies, while others have been retrospective, open-label studies. Studies of sildenafil in PH-LHD have been inconsistent; while some have seen improvements in exercise capacity, haemodynamics and quality of life in patients with systolic and diastolic heart failure [53, 55], others have found no benefit [59, 62].
Completed and ongoing trials of pulmonary arterial hypertension (PAH)-specific therapies in patients with suspected pulmonary hypertension associated with left heart disease (PH-LHD)
There are several limitations to clinical trials for pharmacological agents in PH-LHD. Patients are seldom stratified by the underlying cause of PH, and haemodynamic evaluation is not carried out systematically [23]. It has been recommended that patients with post-capillary PH with a pre-capillary component should represent the target population for clinical trials [4]; however, there are very few current studies in this patient population. In addition, it has been recommended that trial end-points should assess safety first, and efficacy can then be based on measurable clinical outcomes [4]. One of the few trials to address the limitations of previous studies is MELODY-1 [57], an ongoing trial that is evaluating the safety and tolerability of macitentan, an endothelin receptor antagonist, in patients with post-capillary PH with a pre-capillary component. The primary (safety) outcome will be the proportion of patients experiencing significant fluid retention or a worsening of WHO functional class from baseline. The secondary (efficacy) outcomes will be the change from baseline in cardiopulmonary haemodynamic parameters and echocardiographic parameters of systolic and diastolic function after 12 weeks of treatment [57].
In conjunction with specific pharmacological strategies for managing PH-LHD, treating patients' underlying comorbidities is beneficial, e.g. the aggressive control of cardiovascular risk factors [23]. Obesity frequently coexists with PH, and in severe obesity an excess volume load is placed on the left ventricle, which can eventually lead to heart failure [63]. Haemodynamic improvement has been observed with bariatric surgery for PH [64], and this could be explored further as a potential therapy for PH-LHD in patients who have concomitant obesity.
Case study part 2: follow-up
Based on the patient's initial assessment, would you have diagnosed him with PH-LHD?
A volume challenge with 500 mL of normal saline during RHC did not result in an increase in PAWP. Despite his extensive cardiac risk factors and history, it was felt that the PH in this patient was predominantly group 1 PAH. Because of the severity, intravenous epoprostenol was initiated with slow titration, to reach a final initial dose of 6 ng·kg−1·min−1.
Surveillance RHC 1 year later (at an epoprostenol dose of 37 ng·kg−1·min−1) showed a right atrial pressure of 1 mmHg, right ventricular pressure of 71/7 mmHg, systolic/diastolic/mean PAP of 75/36/48 mmHg, PAWP of 12 mmHg, cardiac output/cardiac index of 3.6/2.0 L·min−1, PVR of 799 dyn·s·cm–5 (9.9 Wood units) and systemic vascular resistance of 2069 dyn·s·cm–5 (25.9 Wood units). PVR was decreased by 36%, the PVR/systemic vascular resistance ratio had increased from 0.35 to 0.39, cardiac output had increased by 67% and the patient was WHO functional class II.
Interestingly, 4 years after his diagnosis, the patient's 33-year-old daughter was diagnosed with PAH. Genetic testing in the patient demonstrated a bone morphogenetic protein receptor 2 (BMPR2) mutation. This same mutation was present in the daughter.
Although many of the patient's clinical signs and risk factors were suggestive of PH-LHD, the presence of the BMPR2 mutation also demonstrated the likelihood of heritable PAH. This case, although somewhat atypical, demonstrates that in real life there can be more than one potential cause of PH and that such a scenario of multiple potential aetiologies of PH is not uncommon. In addition, it is important to remember that there is no standard presentation of PH-LHD, and that even cases that appear superficially to be PH-LHD may not be such. Thus, it is important to consider all aspects of the patient's condition. Diagnosing PH-LHD is certainly challenging, with considerable potential for misdiagnosis.
Conclusion
There are multiple unmet needs for patients with PH-LHD. Despite PH being a frequent complication of left heart disease, data on the true incidence are sparse. The differing methodology of epidemiological studies make estimation of incidence challenging, and this is compounded by variations in haemodynamic definitions and terminology, wide variations in disease presentations, and a lack of knowledge surrounding the aetiology and differing phenotypes of PH-LHD. The case presented here highlights the complexity of differentiating PH-LHD from other types of PH, and the multiple factors that should be considered before confirming the diagnosis. Although there is no single pharmacotherapy that is specific for PH-LHD, there are promising signs that clinical trial designs will be adapted in the future to include patients with the most appropriate risk–benefit profile, and to select more relevant clinical end-points.
Acknowledgements
The authors would like to thank Kate Bradford from PAREXEL (Uxbridge, UK) for medical writing assistance, funded by Actelion Pharmaceuticals Ltd (Allschwil, Switzerland).
Footnotes
Conflict of interest: Disclosures can be found alongside the online version of this article at err.ersjournals.com
Provenance: Publication of this peer-reviewed article was sponsored by Actelion Pharmaceuticals Ltd, Allschwil, Switzerland (principal sponsor, European Respiratory Review issue 138).
- Received August 21, 2015.
- Accepted October 14, 2015.
- Copyright ©ERS 2015.
ERR articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References









