





Abstract
Interstitial lung disease (ILD) encompasses a large and diverse group of pathological conditions that share similar clinical, radiological and pathological manifestations, despite potentially having quite different aetiologies and comorbidities. Idiopathic pulmonary fibrosis (IPF) represents probably the most aggressive form of ILD and systemic sclerosis is a multiorgan fibrotic disease frequently associated with ILD. Although the aetiology of these disorders remains unknown, in this review we analyse the pathogenic mechanisms by cell of interest (fibroblast, fibrocyte, myofibroblast, endothelial and alveolar epithelial cells and immune competent cells). New insights into the complex cellular contributions and interactions will be provided, comparing the role of cell subsets in the pathogenesis of IPF and systemic sclerosis.
Abstract
Distinct cell populations contribute to the complex pathogenesis of IPF and systemic sclerosis-associated ILD http://ow.ly/AjFaz
Introduction
The interstitial lung diseases (ILDs) are a group of diffuse parenchymal lung disorders that are classified according to specific clinical, radiological and histopathological features. Often, ILDs have no identifiable underlying causes and are defined as idiopathic [1]. Frequently, however, ILDs can also be associated with a specific environmental exposure or an underlying connective tissue disease.
The lung is susceptible to various forms of short- and long-term injuries, both airborne and blood-borne, that may result in fibrosis. Some forms of fibrosis, such as acute lung injury or cryptogenic organising pneumonia, are at least partially reversible, whereas others, in particular idiopathic pulmonary fibrosis (IPF), are progressive and fatal.
The term idiopathic interstitial pneumonia encompasses a wide range of different ILDs with unique clinical, radiological and pathological features. The examination of surgical lung specimens and an integrated clinico-radiological evaluation allow the start of a complex differential diagnosis and prediction of survival, through the identification of different interstitial patterns [2].
In this setting, IPF is defined as a chronic progressive fibrotic ILD, with unknown aetiology and a pattern of usual interstitial pneumonia (UIP), which is represented by heterogeneous lesions at different stages of evolution with foci of proliferative fibroblasts. IPF is generally considered to be unresponsive to “standard” therapies [3] and has a poor prognosis, with most patients dying within 5 years of diagnosis [4, 5]. In systemic sclerosis (SSc) patients, the change in the trend of scleroderma-related deaths over the past 30 years demonstrates that lung involvement (both pulmonary hypertension and pulmonary fibrosis) is among the primary causes of mortality [6]. In SSc, pulmonary disease can exist as a heterogeneous combination of pulmonary fibrosis and pulmonary arterial hypertension (PAH), but the pathogenic mechanisms that induce different pulmonary responses are poorly understood. Although SSc-associated ILD (SSc-ILD) is associated more frequently with a nonspecific interstitial pneumonia pattern, which is characterised by homogeneous lesions with predominant inflammatory infiltrates, some aspects of IPF and SSc-ILD suggest that these diseases could share some common features [7].
Lung fibrotic disorders are characterised by accumulation of fibroblasts, myofibroblasts and extracellular matrix (ECM), leading to chronic respiratory failure. The origins of the fibroblasts and their activation remain debated and are probably multiple (fig. 1) [8]. The respective roles of the proliferation of resident fibroblasts or mesenchymal precursors, of the transition of epithelial, endothelial or mesothelial cells towards a mesenchymal phenotype, of the recruitment of circulating blood mesenchymal precursors and of the contribution of the coagulation cascade and the activation of the immune system will be discussed in this review, highlighting shared cellular mechanisms and their interactions that are involved in IPF and in SSc-ILD pathogenesis (fig. 2).
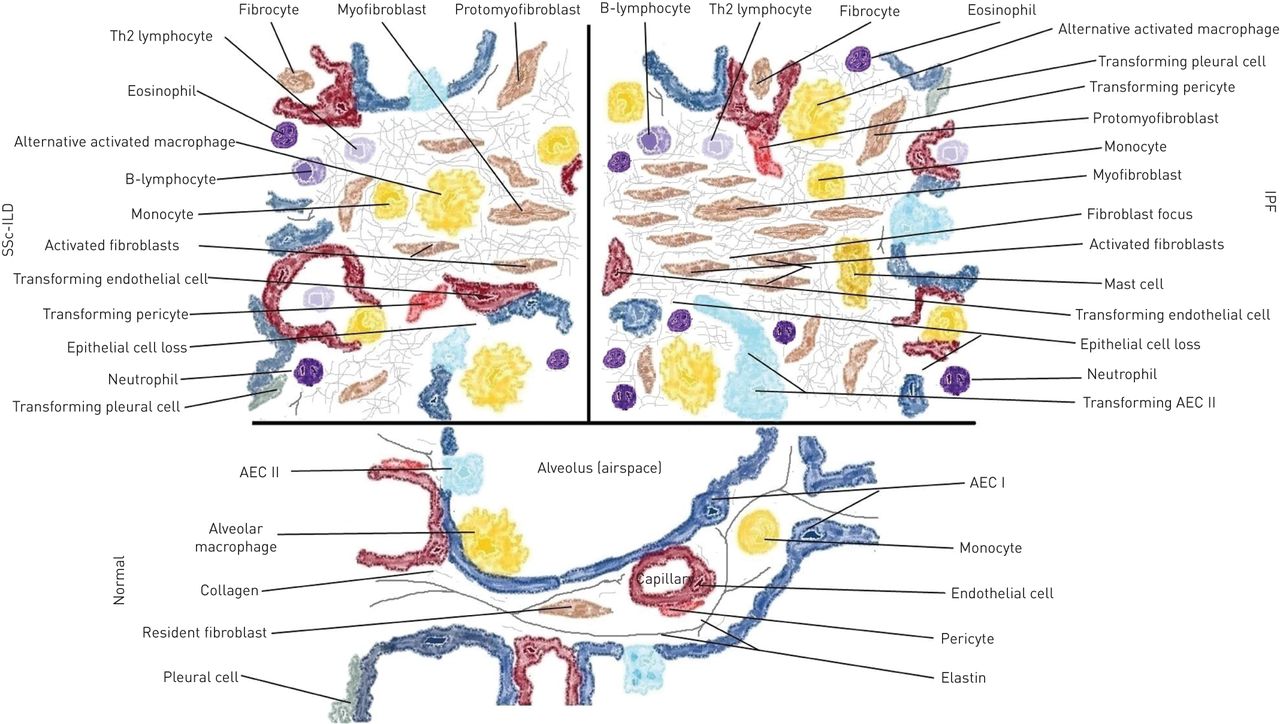
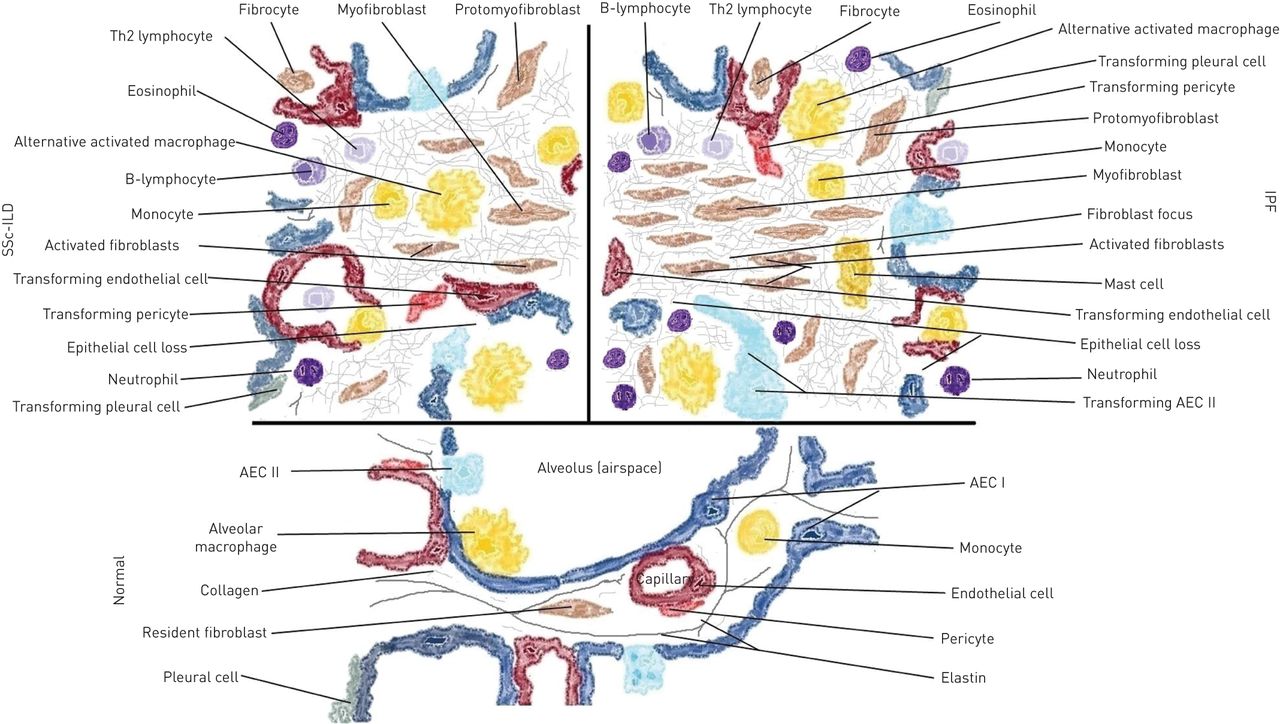
This schematic view of the morpho-functional unit of the lung (alveolus) depicts the main differences in cellular composition in idiopathic pulmonary fibrosis (IPF) and systemic sclerosis-associated interstitial lung disease (SSc-ILD) compared with normal physiological cellular components. Th2: T-helper cell type 2; AEC: alveolar epithelial cell.
{kind=link}
{kind=link}
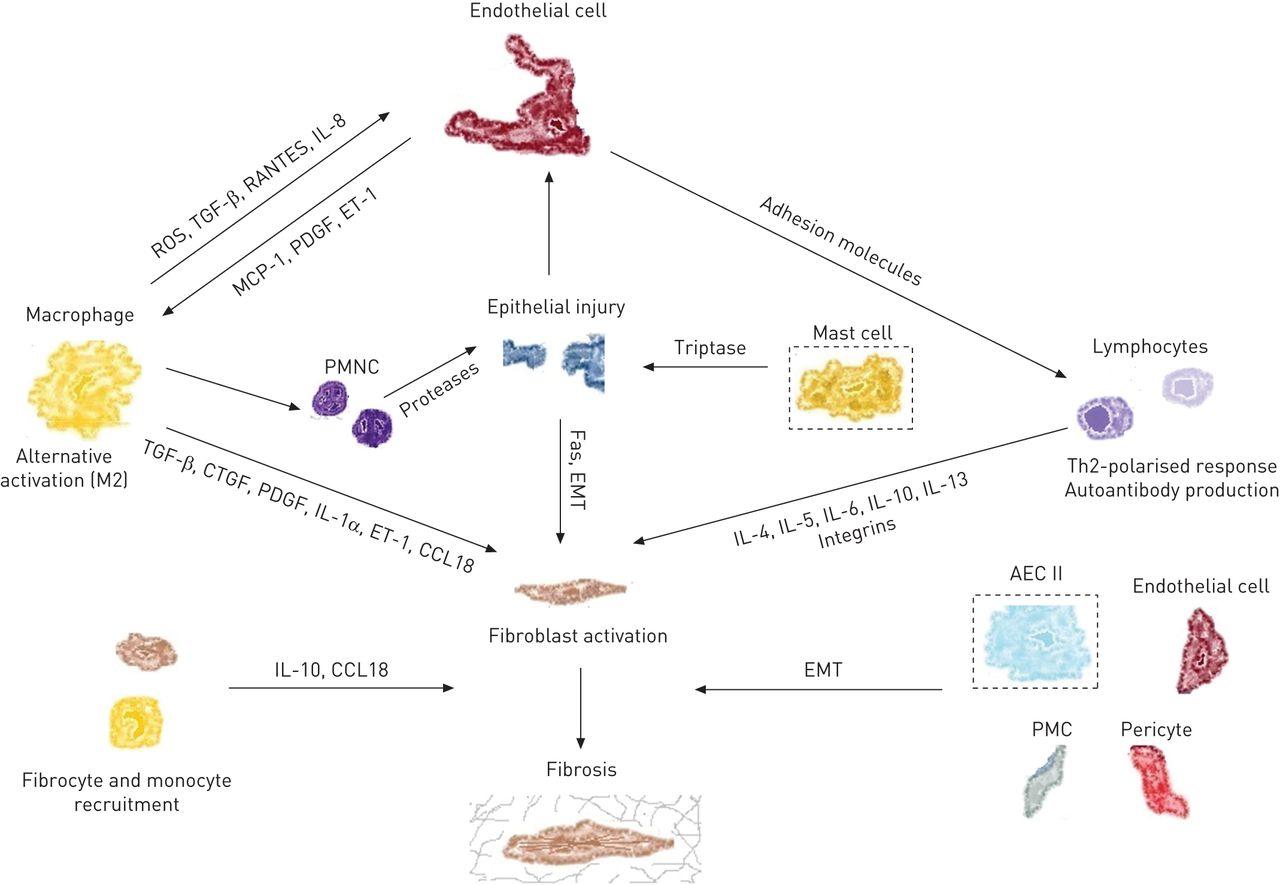
Cellular players and molecules in idiopathic pulmonary fibrosis (IPF) and systemic sclerosis-associated interstitial lung disease. Cells inside dashed lines are relevant to IPF pathogenesis only. ROS: reactive oxygen species; TGF: transforming growth factor; IL: interleukin; MCP: monocyte chemotactic protein; PDGF: platelet-derived growth factor; ET: endothelin; PMNC: peripheral blood mononuclear cell; EMT: epithelial–mesenchymal transition; CTGF: connective tissue growth factor; Th2: T-helper cell type 2; AEC: alveolar epithelial cell; PMC: pleural mesothelial cell.
Myofibroblasts: the effector cells of fibrosis
Myofibroblasts are thought to play a major role in fibrosis through excessive deposition of ECM. These spindle- or stellate-shaped cells share features with smooth muscle cells in that they are contractile and contain α-smooth muscle actin (α-SMA) stress fibres. Myofibroblasts regulate ECM remodelling by combining the synthesising features of fibroblasts with cytoskeletal contractile characteristics of smooth muscle cells [9].
The lung contains more than 40 different cell types, yet most of the increased deposition of ECM found in IPF is due to the activation of myofibroblasts in fibroblast foci, the histopathological hallmarks. These lesions consist of aggregates of activated fibroblasts that produce excessive levels of ECM within the alveolar space at the site of epithelial cell loss. Importantly, they do not arise in healthy lungs, their number correlates with survival [10] and profusion of fibroblastic foci is the most discriminative feature for separating idiopathic from collagen vascular disease-associated UIP. In SSc, the presence of myofibroblasts is prominent in areas with collagen deposition, such as the skin and the lung, but they are also observed in the oesophagus and even in the liver, despite the absence of fibrosis. Culture of bronchoalveolar lavage (BAL) fluids from patients with scleroderma shows spontaneous outgrowth of α-SMA-positive cells with high production of collagen and fibronectin, while in contrast, myofibroblasts are not detected in BAL fluids from healthy individuals. This difference suggests that SSc myofibroblasts possess the ability to maintain the features of ECM-producing cells and/or stimulate each other also outside fibrotic areas and the associated biochemical milieu [11].
It is well demonstrated that myofibroblasts have multiple origins (table S1) and contribute significantly to tissue remodelling by exerting traction forces and synthesising ECM components [12]. Recent in vitro studies demonstrate that myofibroblasts use a lock-step mechanism of cyclic and incremental contractile events based on different ranges of contractions mediated by RhoA/Rho-associated kinase and by changes in intracellular calcium concentrations [13].
Despite the fact that the origins of myofibroblasts may be very heterogeneous, their development follows a well-established sequence of events after tissue injury: in response to mechanical challenge, fibroblasts acquire contractile stress fibres that are first composed of cytoplasmic actins, conferring the features of the protomyofibroblast, which represents an intermediate phenotype preceding the differentiated myofibroblast that is characterised by de novo expression of α-SMA [14].
Myofibroblasts have at least three possible origins. The first hypothesis postulates that resident fibroblasts differentiate into myofibroblasts after multiple profibrotic stimuli. The second addresses the possibility that circulating fibrocytes or other bone marrow-derived progenitor cells could be recruited to the site of injury. A third possible source of fibroblasts and/or myofibroblasts in pulmonary fibrosis could be constituted by the differentiation of various cell subsets through the process of mesenchymal transition [15].
Fibroblasts: resident cells, apoptosis and the transition to myofibroblasts
Fibroblasts are widely distributed in all lung structures and play a key role in matrix homeostasis. Fibroblasts modulate ECM turnover through the expression of matrix metalloproteinases, which degrade ECM, and their inhibitors, tissue inhibitors of metalloproteinases (TIMPs). Fibrosis is induced by activation, proliferation and migration of these cells into the site of injury and deposition of matrix proteins. Fibroblasts have different patterns of differentiation related to the site where they are located and, despite the factors that determine their positional identity remaining unclear, visceral and cutaneous fibroblasts have different transcriptional programmes [16].
Fibroblasts are key players in the pathogenesis of IPF and SSc-ILD. Recently, Hsu et al. [17] compared gene expression of lung fibroblasts of patients affected by SSc-associated pulmonary fibrosis (SSc-PF) and SSc-associated PAH to IPF and idiopathic PAH. SSc-PF and IPF lungs and fibroblasts express genes involved in fibrosis, such as collagen types I and III, TIMP1 and interferon (IFN)-γ receptor, and genes involved in insulin-like growth factor (IGF) signalling, such as IGF-binding proteins (IGFBPs), secretory leukocyte peptidase inhibitor and connective tissue growth factor (CTGF). Proteins with high affinity for IGF, such as IGFBP-3 and IGFBP-5, as well as low-affinity IGFBPs, such as IGFBP-7, CTGF and CYR61 (cysteine-rich angiogenic inducer 61), were also aberrantly expressed in fibrotic lungs and fibroblasts from IPF and SSc-ILD [17].
Among these, IGFBPs seem to have a prominent role in the regulation of fibrosis. IGFBP-3 independently induces syndecan-2 (SDC2) and regulates transforming growth factor (TGF)-β induction of SDC2 in primary human fibroblasts from IPF and SSc-PF patients [18].
Egr-1 (early growth response protein 1) expression is also increased in lesional skin and lung biopsies from patients with diffuse SSc [19] and lungs of patients with IPF [20]. Egr-1, a zinc finger transcription factor that is upregulated in response to TGF-β, appears to be required for physiological and pathological connective tissue remodelling, regulates collagen gene expression and promotes fibrotic gene transcription together with IGFBP-5 [21].
In contrast to epithelial and endothelial cells, fibroblasts become resistant to apoptosis, both in IPF and in SSc. Thannickal and Horowitz [22] suggested that this “apoptosis paradox” is central to the pathogenesis of IPF. In fact, experimental studies on IPF show that inhibitors of the pro-apoptotic molecules inhibit fibrosis, confirming that epithelial cell apoptosis is an important event [23]. Moreover, the expression of mediators of cell death, such as caveolin-1 (cav-1) and Fas proteins, is low in fibroblasts within the fibroblastic foci of IPF patients, suggesting that another pathway pathologically altered is the PTEN (phosphatase and tensin homologue)/Akt axis, which inactivates Fox (forkhead box) O3a, downregulating cav-1 and Fas expression [24]. This confers to IPF fibroblasts a different phenotype, resistant to apoptosis, and may be responsible for IPF progression. A central role for cav-1 in the development of pulmonary fibrosis is further supported by recent evidence showing that cav-1 is markedly decreased in affected lungs and skin from SSc patients and cav-1 knockout mice develop dermal and pulmonary fibrosis [25].
Fibroblasts in most tissues vary with respect to size, secretory profile and surface markers and, within a fibrogenic milieu, are clearly different from those in normal tissues [26]. Indeed, cell–matrix interactions are active areas of investigation, in particular the activation of focal adhesion kinase (FAK), since it has been shown that multiple stimuli, acting on distinct cell surface receptors, converge to induce a common response, the tyrosine phosphorylation of FAK [27]. Furthermore, the constitutive activation of SSc myofibroblasts requires, at least partly, FAK phosphorylation [28], and the administration of FAK inhibitors regulates pulmonary fibrosis in bleomycin-treated animals [29].
Wnt/β-catenin signalling is another common area of interest between SSc-ILD and IPF [30, 31]. The inhibition of the Wnt/β-catenin signalling pathway is associated with lung fibroblast activation, differentiation and the dysregulation of repair processes [32]. Confirmatory results come from gene profiling studies, and show that sFRP-1 (secreted frizzled-related protein 1), an antagonist of the Wnt signalling pathway, is decreased in fibroblasts from fibrotic lungs irrespective of the fibrotic disease [17].
In conclusion, the underlying mechanisms responsible for the switch to the activated status of fibroblasts remain poorly explained. One possible scenario is that other cells present in the fibrotic lesion during early stages, e.g. inflammatory cells, produce the signals that either “activate” the resident fibroblasts or selectively amplify the subpopulation of fibroblasts with the pre-existing activated phenotype. The constitutive activation of SSc fibroblasts probably reflects their persistent autocrine stimulation by TGF-β and other profibrotic stimuli [33]. TGF-β, acting in concert with other cytokines or chemokines, may constitute such signals. In addition, cell–cell contact between immune cells or alveolar epithelial cells (AECs) and fibroblasts may play a role in fibroblast activation and/or selection.
Fibrocytes: circulating precursors of fibroblasts, collagen-producing monocytes and recruitment to the site of injury
Fibrocytes are mesenchymal progenitors derived from haematopoietic precursors, identified by the co-expression of the stem cell marker CD34, the leukocyte common antigen CD45 and fibroblast markers (collagen 1+), and implicated in a wide variety of focal and diffuse remodelling disorders [34]. In vitro and in vivo experiments confirm that the inhibition of fibrocyte recruitment or the blockade of their differentiation from precursors limit the development of lung fibrosis [35]. Multiple studies also demonstrate that elevated fibrocyte levels are found in the blood and in the lungs of patients with IPF [36] and SSc-ILD [37]. However, the association between disease severity and the increased number of circulating fibrocytes has not been unequivocally confirmed [38].
Fibrocytes express on their surface the C-X-C chemokine receptor type 4 (CXCR4), which mediates the migration of these cells in response to stromal cell-derived factor 1 (CXCL12). The expression levels of CXCR4 and its ligand, CXCL12, are highly upregulated in SSc-ILD [37] and IPF lung tissue [39]. In summary, these data suggest that the blockade of fibrocyte recruitment to the site of injury could represent a potential therapeutical target [40].
AECs: pivotal cells in IPF and their role in epithelial–mesenchymal transition
Traditionally, IPF was considered a chronic inflammatory-driven response caused by the abnormal accumulation of inflammatory cells, such as alveolar macrophages and neutrophils, and by decreased capacity of fibroblasts to synthesise the anti-inflammatory and antifibrogenic molecule prostaglandin (PG)E2 [41], but current evidence suggests that the fibrotic response could also be driven by abnormally activated AECs.
In IPF it is hypothesised that the injury occurs to the lung epithelium, probably to type I AECs that line the majority of the alveolar surface [42]. When type I AECs are damaged, type II AECs undergo hyperplastic proliferation to protect the injured basement membranes. In normal repair, the hyperplastic type II AECs will undergo regulated apoptosis. The remaining cells will spread and undergo a differentiation process to become type I AECs through the Smad-dependent TGF-β1 pathway. Under pathological conditions, fibroblasts migrate into these areas of damage and differentiate into myofibroblasts, which initiates the fibrotic process. Based on the evidence that targeted deletion of type II AECs leads directly to fibrosis [43], and on the existence of human genetic diseases in which mutations of surfactant protein C [44] lead directly to endoplasmic reticulum stress, type II AEC death and pulmonary fibrosis, injured AECs are viewed as potential drivers of pulmonary fibrosis.
There are three different theories under discussion regarding the mechanisms through which injury to the type II AECs results in lung fibrosis. The first confers to epithelial cells the ability to undergo transdifferentiation into fibroblasts and consecutive activation through the process of epithelial– mesenchymal transition (EMT). While confirmatory results have been obtained on this process from experiments in mice [45], the data are controversial regarding the importance of this mechanism in IPF patients [46, 47]. Indeed, it has been demonstrated that AECs can differentiate into myofibroblasts in vivo during experimental fibrosis and ex vivo in response to TGF-β1 and Wnt signalling, through Smads and β-catenin, respectively. Cell–matrix interactions could further reinforce this transition mechanism as evidenced by other experimental data on integrin α3β1 [48] and αvβ6 [45]. Abnormal alveolar fibrin turnover has been reported to play a role in the development of pulmonary fibrosis and has been involved in alveolar EMT: coagulation proteases may exert profibrotic cellular effects via protease-activated receptors (PARs) that can support thrombin-induced EMT. This provides the first evidence that PAR-3, via its ability to potentiate thrombin-triggered EMT, could potentially contribute to the pathogenesis of pulmonary fibrosis via multiple mechanisms (table S1) [49]. In particular, an altered balance between the activating enzyme urokinase-type plasminogen activator (uPA) and its inhibitor plasminogen activator inhibitor (PAI)-1 seems to contribute to the development of pulmonary fibrosis. Recently, Manetti et al. [50] demonstrated that uPA receptor-deficient mice closely resemble the histopathological features of SSc, including tissue fibrosis and peripheral microvasculopathy. Moreover, plasminogen activation augments PGE2 secretion, which represents an antifibrotic signal, in fibroblasts and also in fibrocytes and AECs. PGE2 production is also stimulated by the activation of the hepatocyte growth factor (HGF)/cyclooxygenase (COX)-2/PGE2 axis [51]. Theoretically this, in turn, should contribute to an antifibrotic milieu by the ability of HGF to promote epithelial cell survival and to inhibit epithelial–mesenchymal transdifferentiation through Smad7 activation [52]. Despite a series of studies that have shown that HGF suppresses fibrosis by inhibiting collagen production, inducing myofibroblast apoptosis and degrading the ECM [53], patients with SSc have increased circulating levels of HGF compared with those in healthy controls [54], probably suggesting that an intracellular defect of pro-HGF pathway activation is present in SSc fibroblasts as encountered in IPF [55].
The second theory proposes that the dying type II AECs lose control over the mesenchymal cells and, as a result, they proliferate and produce more collagen. In detail, PGE2 has been shown to be a key factor controlling fibroblast differentiation and proliferation. In IPF, the reduction of PGE2 levels and, therefore, the loss of control of fibroblastic proliferation due to excessive epithelial apoptosis, may be relevant. In this setting, lysophosphatidic acid (LPA)1 receptor has been gaining increasing interest, since it has been recently demonstrated that it can induce apoptosis of bleomycin-treated epithelial cells [56]. Moreover, LPA levels are increased in the broncoalveolar lavage of IPF patients [57] and in the serum of patients affected by SSc-ILD [58].
A third putative mechanism may be that the dying type II AECs release factors such as stromal cell-derived factor 1 (CXCL12), attracting circulating fibrocytes, which in turn invade the lung and locally expand the fibroblast pool.
Experiments in aged mice further confirmed that epithelial cells were more likely to undergo endoplasmic reticulum stress and apoptosis [59] and fibroblasts from aged mice overexpressing TGF-β receptors tend to be resistant to apoptosis. Growing evidence suggests that oxidative stress is one of the causes of AEC damage and AEC apoptosis in IPF [60]. An animal model based on reactive oxygen species injury has been established for SSc [61], closely resembling the vascular, fibrotic and autoimmune features of the disease, and increased oxidative stress has been shown both in IPF [62] and in SSc [63].
Endothelial cells: vascular damage, EMT and progenitors
The alterations of vascular function and microcirculatory abnormalities are the earliest clinical manifestations of SSc, and may precede the onset of fibrosis by months or years [64]. The vascular damage in SSc primarily affects the microvasculature (small and medium-sized arteries, arterioles and capillaries) and can be observed in all affected organs. The LPA1 receptor, which regulates the endothelial barrier function, has been shown to drive pulmonary fibrosis in murine models of lung fibrosis, supporting a link between vascular damage and the development of fibrosis. However, the LPA-mediated loss of barrier function in human pulmonary artery endothelial cells appears to be independent of the LPA1 receptor and probably mediated by the LPA6, LPA2 and LPA4 receptors [65]. In human IPF, the imbalance between angiogenic and angiostatic factors influences the vascular rarefaction seen with the progression of the damage: the reduced expression of angiogenic factors, such as vascular endothelial growth factor (VEGF), is paralleled by elevation of angiostatic molecules, such as pigment epithelium-derived factor [66, 67].
Blood vessels in the lung originate from existing vessels through angiogenesis (proliferation and migration of existing endothelial cells), but it has been shown that vasculogenesis (new development of vessels by stem or progenitor cells) may be important for the homeostasis of peripheral arteries and capillaries [68]. Vasculogenesis was long regarded to be related to embryogenic processes, but the discovery of endothelial progenitor cells (EPCs) in adult bone marrow and peripheral blood has proved that vasculogenesis is active throughout life [69]. EPCs represent a subset of bone marrow-derived stem cells that play an essential role in vascular repair. Two subtypes of EPCs exist: late EPCs may differentiate into mature endothelial cells and repair injured blood vessels, while early EPCs (i.e. EPCs that grow into colony-forming units on fibronectin following 5–7 days of culture) may have angiogenic potential by secreting cytokines such as VEGF and enhance the angiogenic process. Early EPC levels are reduced in IPF patients compared with controls, and increased endogenous VEGF expression in early EPCs derived from IPF patients may reflect a compensatory mechanism to overcome reduced EPC levels and retain endothelial homeostasis in the IPF lung [70]. Data on EPCs in SSc are controversial: some reports suggest that EPC levels are decreased regardless of SSc stage [71], while other authors demonstrated increased EPC levels in the early/active stage and subsequent reduction with disease progression and severity [72]. Certainly the ability of these bone marrow-derived CD133+ cells to differentiate into endothelial cells in vitro is impaired in SSc [73], and probably represents a functional defect in this cell subset, which contributes to endothelial dysfunction and defective repair.
Although a few in vitro studies have reported the possibility of endothelial cells being a source of myofibroblasts [74], it has not been fully determined whether, in humans, endothelial cells could undergo EMT under certain pathological conditions such as lung fibrosis. However, it has been demonstrated that endogenous lung endothelial cells give rise to significant numbers of fibroblasts in a murine model of bleomycin-induced lung injury and fibrosis [75].
Pericytes and pleural mesothelial cells: underestimated contributors to mesenchymal transition
Lung pericytes are mesenchyma-derived cells that are localised within basal membranes or perivascular linings [76] and are involved in wound healing and collagen production [77]. More than 20 years ago, this cell type was considered a source of myofibroblasts in other fibrotic processes [78], and it has been demonstrated that pericytes in fibrotic lungs are no longer confined by the capillary basement membrane [79]. In SSc, the role of pericytes has been investigated mostly regarding skin involvement, and it is likely that a cell type-specific marker of pericytes and scleroderma myofibroblasts, the regulator of G-protein signalling (RGS)5, may provide a key link between initial hyperplasia and fibrosis in this disease [80]. However, the specific contribution of pericytes to pulmonary fibrotic disease needs to be better elucidated.
The pleura is largely constituted by mesothelium, which originates from the embryonic mesoderm and consists of flattened, squamous-like epithelial cells that are surrounded by dense connective tissue. Pleural mesothelial cells (PMCs) have been implicated in the process of transformation into myofibroblasts and migration, both in vitro [81] and in vivo [82], through a TGF-β1-dependent mechanism called mesothelial–mesenchymal transition. PMCs are present in IPF lungs, and their number correlates with IPF severity [83]. The specific role of PMCs in SSc-ILD needs to be investigated further.
Macrophages: monocyte recruitment, alternative activation and defective efferocytosis
Immune cells appear to play a pivotal role in the development of both SSc-ILD and IPF (table 1). In the lungs of SSc patients with ILD, macrophages are predominant but T-cells are also a major constituent in most patients [84].
Immune cells in idiopathic pulmonary fibrosis (IPF) and systemic sclerosis-associated interstitial lung disease (SSc-ILD)
Macrophages are a prominent cell type in pulmonary fibrotic processes [85] and become profibrotic through alternative activation by interleukin (IL)-13. A paradigm for macrophage activation has been defined on the basis of cytokines released by the two T-helper cell (Th) subtypes. “Classically activated” macrophages are activated by the Th1 cytokine IFN-γ, whereas “alternatively activated” macrophages are activated by the Th2 cytokines IL-4 and IL-13, but also by glucocorticosteroids and ingestion of apoptotic material, acquiring an anti-inflammatory, phagocytic, profibrotic phenotype [86]. They are also identified in part by the expression of the scavenger receptor CD163, the IL-1RII decoy receptor and mannose receptors, and by increased IL-10, C-C chemokine ligand (CCL)18 and pulmonary activation-regulated chemokine (PARC) production [87]. Different ILDs, including IPF and SSc-ILD, share the increased production of CCL18, a chemokine produced mainly by antigen-presenting cells that is chemotactic predominantly for T-lymphocytes, as a common mechanism, and CCL18 produced by alveolar macrophages promotes collagen production by lung fibroblasts [88]. The major source of both mouse and human IL-10 is activated monocytes, but B-cells and type II AECs [89] also produce this cytokine and levels of IL-10 are significantly elevated in both IPF and SSc-ILD [37]. Accumulating data demonstrate that monocyte-derived cells in the lungs and circulation of humans with many forms of pulmonary fibrosis exhibit discrete profibrotic phenotypes characterised by expression of markers of alternative macrophage activation [37]. CD14+ monocyte precursors obtained from the peripheral blood exhibit a similar alternative activation phenotype when stimulated by the Toll-like receptor (TLR)4 agonist lipopolysaccharide (LPS) [90]. Because LPS is traditionally associated with classical activation, these data indicate that monocytes from patients with SSc-ILD may be preprogrammed to adopt an alternative activation state. In addition, TLR4 and innate immune signalling may be another important area of investigation in SSc-ILD [91]. A number of mediators associated with alternative activation, such as IL-13, IL-1 receptor antagonist, CCL17, CCL2, CXCL8, CCL22 and S100A9, are associated with pulmonary fibrosis [92], together with other TLRs such as TLR2 [93] and TLR9 [94]. Nevertheless, macrophages can secrete a variety of profibrotic mediators, including platelet-derived growth factor, TGF-β [95] and reactive oxygen species [96]. Therefore, it appears that the alternative activation of macrophages exerts a crucial step in the evolution of pulmonary fibrosis in both IPF and SSc-ILD.
Lymphocytes: Th2-polarised response and antibody production
Various pulmonary fibrotic diseases are frequently associated with excessive accumulation of T-lymphocytes. T-lymphocytes are present diffusely throughout the lungs, with focal perivascular aggregates and within alveolar spaces. T-lymphocytes represent a relatively small population in a normal lung; however, they accumulate when pulmonary inflammation and fibrosis occur [97].
An abnormal profibrotic Th2-polarised T-cell response has been postulated to mediate tissue damage and fibrosis in SSc-ILD [98] and in IPF [99]. Analyses of the expression of CXCR3 and CCR4 (chemokine receptors associated in vitro with Th1 and Th2 cells, respectively) suggest a dominance of Th2 cells in SSc-ILD and IPF cases [100, 101].
In this setting, a previously unrecognised role for integrin-mediated activation of T-cells in the regulation of pulmonary inflammation and fibrosis has been recently shown in SSc patients. In vitro experiments revealed that T-cells overexpressing the integrins αvβ3 and αvβ5 are directly profibrotic on cultured primary human pulmonary fibroblasts, probably through a TGF-β-dependent mechanism [102].
Decreased numbers of regulatory T-cells (Tregs) expressing CD4 and FoxP3 have been described in the lungs and circulation of patients with IPF [103], while in SSc patients the same cell population has been reported to be increased, despite the fact that production of Treg mediators, such as TGF-β1 and IL-10, was unmodified, suggesting possible alterations in function [104]. SSc-ILD was strongly associated with higher numbers of IL-22-producing lymphocytes, while controversial results exist for Th17 [105].
B-cells have also been implicated in the pathogenesis of pulmonary fibrosis, either by producing autoantibodies or by secreting IL-6 [106]. In IPF, plasma B-lymphocyte stimulator (BLyS) concentrations correlate with poor prognosis and disease manifestations [107]. Experimental data demonstrated that treatment with anti-topoisomerase I antibody induces skin and lung fibrosis stimulating IL-6 production [108]. Moreover, IL-6 is an independent predictor of decline in diffusing capacity of the lung for carbon monoxide (DLCO), in both IPF and SSc-ILD patients [109]. Oncostatin M, an IL-6 family cytokine, has been implicated in a number of biological processes, including the modulation of ECM and the induction of pulmonary inflammation and fibrosis, both in SSc-ILD and IPF [110], through an IL-4/IL-13 and TGF-β-independent mechanism.
Anti-topoisomerase I (anti-Scl-70) antibodies are specific antibodies for SSc and have been associated with a poor prognosis and higher mortality rate [111]. Many other autoantibodies have been detected in SSc patients and a link between them and severity of SSc-ILD has been established for anti-endothelial cell antibodies [112], for autoantibodies to fibroblasts that have been shown to induce an activated fibroblast phenotype responsible for ECM deposition in lungs [113], and for autoantibodies against phosphatidylserine-prothrombin complex [114]. In IPF, the presence of autoantibodies against DNA topoisomerase IIα, another form of topoisomerase able to induce transient double-stranded breaks in the DNA, has been shown in the sera of one-third of patients with IPF [115].
Further confirmatory data on the involvement of lymphocytes come from the demonstration of improved lung function in SSc-ILD patients treated with the lymphocyte-modulating agents mycophenolate [116] or rituximab [117].
Natural killer (NK) cells are innate immune cells that produce various cytokines. A higher number of NK cells has been reported in the BAL and in the blood of patients affected by IPF and SSc [118, 119]. Increased levels of several cytokines have been demonstrated in nonstimulated NK cells from SSc patients when compared with controls, suggesting that NK cell dysfunction may contribute to immunological abnormalities in scleroderma.
Mast cells: fibrogenesis and lung infiltration
Traditionally, mast cells have been recognised as the effectors of IgE-mediated allergic inflammation. More recently, their role has been highlighted in other disease processes such as fibrogenesis [120]. Mast cells and fibroblasts share common residence in the mesenchymal compartment, and their membranes are also tethered in close apposition in fibrotic lung tissues [121]. Several studies have suggested that human mast cells are a potential source of profibrotic factors such as TGF-β [122], and that tryptase stimulates lung fibroblast growth in a PAR-2/protein kinase (PK)C-α/Raf-1/p44/42-dependent manner and potentiates ECM production [123]. Nonetheless, it seems that they play no role in development of lung fibrosis following bleomycin-mediated lung injury [124], and recently it has been demonstrated that mast cell density is increased in IPF compared with SSc-ILD and is related inversely to disease progression [125].
Polymorphonuclear cells: inflammation and reactive oxygen species injury
Polymorphonuclear cells have long been recognised to infiltrate the lungs of patients affected by IPF [126], yet still the contribution of these cells in the pathogenesis of the disease is a matter of debate [127]. Their role has been associated with the persistent production of elastases and reactive oxygen species and the amplification of lung damage through the production of profibrotic cytokines. Neutrophil recruitment is also an important predictor of early mortality in IPF patients [128]. BAL studies have identified that these populations of inflammatory cells are present at high levels in the lungs of patients with IPF compared with SSc-ILD [129], and their number correlates with disease severity [130, 131]. Recently, it has been demonstrated that BAL neutrophil and eosinophil cell counts are directly correlated with the concentration of CCL18 [132], which was in turn inversely correlated with total lung capacity and the DLCO in both disease groups [132]. Experimental studies, using both genetic and chemical models, suggest that the blockade of elastase is beneficial in pulmonary fibrosis animal models [133]. Although there is an evident contradiction between the high infiltration of polymorphonuclear cells in the lungs of patients affected by pulmonary fibrosis and the disappointing results obtained by the use of anti-inflammatory agents [134], it would appear necessary to further elucidate the role of these cells in the pathogenesis of IPF and SSc-ILD and to find specific cell-targeted therapies.
Conclusion
Distinct cell populations seem to contribute to the complex and diverse pathogenesis of pulmonary fibrosis. Although IPF and SSc-ILD are different conditions in terms of clinical and radiological features and response to therapy, it appears that there are similarities in cellular transition, plasticity and recruitment, cell–cell interplay, cell–matrix interactions and immune system activation. A great need exists for new disease models, such as the “organ on a chip” platform [135], to study specific cellular responsiveness and to test new potential treatments for these conditions, whose prognosis remains extremely poor. In the future, the acquisition of advanced methods to analyse and modulate profibrotic cellular activation, especially in early stages of the disease, will allow us to discover cell-specific targets and support novel therapeutic strategies for chronic pulmonary diseases.
Footnotes
This article has supplementary material available from err.ersjournals.com
Conflict of interest: None declared.
Provenance: Submitted article, peer reviewed.
- Received April 27, 2014.
- Accepted June 15, 2014.
- Copyright ©ERS 2015.
ERR articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










Jump To
- Article
- Abstract
- Abstract
- Introduction
- Myofibroblasts: the effector cells of fibrosis
- Fibroblasts: resident cells, apoptosis and the transition to myofibroblasts
- Fibrocytes: circulating precursors of fibroblasts, collagen-producing monocytes and recruitment to the site of injury
- AECs: pivotal cells in IPF and their role in epithelial–mesenchymal transition
- Endothelial cells: vascular damage, EMT and progenitors
- Pericytes and pleural mesothelial cells: underestimated contributors to mesenchymal transition
- Macrophages: monocyte recruitment, alternative activation and defective efferocytosis
- Lymphocytes: Th2-polarised response and antibody production
- Mast cells: fibrogenesis and lung infiltration
- Polymorphonuclear cells: inflammation and reactive oxygen species injury
- Conclusion
- Footnotes
- References
- Figures & Data
- Info & Metrics