





Abstract
Comprehensive, up-to-date review of RA-associated lung diseases including pathogenesis and management http://ow.ly/FBaNZ
Introduction
Rheumatoid arthritis is a systemic inflammatory disorder that most commonly affects the joints, causing progressive, symmetric, erosive destruction of cartilage and bone, which is usually associated with autoantibody production. Rheumatoid arthritis affects ∼1% of the population in developed countries. The incidence and prevalence of rheumatoid arthritis in developing countries is thought to be lower, but is difficult to quantify. Although joint disease is the main presentation, there are a number of extra-articular manifestations including subcutaneous nodule formation, vasculitis, inflammatory eye disease and lung disease. Of these manifestations, lung disease is a major contributor to morbidity and mortality. In some cases, respiratory symptoms may precede articular symptoms. It is critical for the pulmonologist to assess for systemic and articular signs and symptoms of connective tissue disease when evaluating a patient with pulmonary disease of unknown aetiology as patients may initially present with pulmonary symptoms. There are a variety of pulmonary manifestations of rheumatoid arthritis, including pulmonary parenchymal disease (interstitial lung disease (ILD)) and inflammation of the pleura (pleural thickening and effusions), airways and pulmonary vasculature (vasculitis and pulmonary hypertension). These changes may reflect chronic immune activation, increased susceptibility to infection (often related to immunomodulatory medications) or direct toxicity from disease modifying or biological therapy. Prognosis varies depending on the type and severity of involvement. Herein, we review the various manifestations of rheumatoid arthritis-associated lung disease, as well as the recent advances in treatment.
Types of pulmonary involvement
Respiratory symptoms in rheumatoid arthritis can be due to a variety of conditions that affect the parenchyma, pleura, airways or vasculature (table 1). Complications may arise directly from rheumatoid arthritis involvement or may occur secondary to immune-modulating medications used to treat rheumatoid arthritis. The majority of respiratory manifestations occur within the first 5 years of disease [1]. Respiratory symptoms may precede onset of articular symptoms in 10–20% of cases [2]. However, they may be masked by poor functional status from joint disease or chronic inflammation.
Pulmonary manifestations of rheumatoid arthritis
Interstitial lung disease
ILD is the most common pulmonary manifestation of rheumatoid arthritis lung disease [3, 4], although the exact prevalence varies depending on the population studied and the diagnostic modality used to define the disease. In an Australian cohort of rheumatoid arthritis patients with a disease duration <2 years, 58% of these patients had changes consistent with ILD on either chest radiograph, high-resolution computed tomography (HRCT), pulmonary function testing (PFT), bronchoalveolar lavage (BAL) and/or 99Tc-DTPA (technetium-99 m-labelled diethylenetriamine pentaacetate) scan. Of these patients, 76% had clinically silent disease [5]. A more recent study of 40 patients, also with <2 years of disease, found that abnormalities on HRCT scans and/or PFTs were present in 45%, with 10% having clinically significant disease [6]. It is currently estimated that ∼30% of patients with rheumatoid arthritis have subclinical ILD noted on HRCT scans [3, 4]. While the rate of some extra-articular manifestations of rheumatoid arthritis have decreased with improvements in therapy, the incidence of ILD has remained fairly stable [7, 8], if not increased [9]. Whether this reflects an increase in detection or is the result of drug-induced lung disease with more aggressive use of anti-rheumatic agents is not entirely clear.
Epidemiology/risk factors
Although rheumatoid arthritis is more common in females, rheumatoid arthritis associated-ILD (RA-ILD) occurs more frequently in males, with a male to female ratio as high as 2:1 in some studies [7, 10]. Onset of lung disease typically occurs in the fifth to sixth decade of life. The incidence of RA-ILD may increase as newer agents allow better disease control and, consequently, increased life expectancy [11]. Age has consistently been shown to be a risk factor for the development of ILD [12]. Another major risk factor is a history of smoking [12], with one study finding an odds ratio of 3.8 for those who smoked >25 pack-years [13]. High levels of rheumatoid factor are a known risk factor for extra-articular manifestations of rheumatoid arthritis, including ILD. The exact mechanism for this has not been elucidated, but may involve formation of circulating immune complexes [14].
Pathogenesis
The mechanism of pulmonary fibrosis occurring in ILD is not well understood (fig. 1). Patients with rheumatoid arthritis typically have circulating autoantibodies, the most common being rheumatoid factor and anti-cyclic citrullinated peptide (CCP). These antibodies may be present in the serum for several years before clinical disease onset [15, 16]. Both rheumatoid factor and anti-CCP have been linked to the development of ILD, particularly when present in high titres [4, 17–21]. Anti-CCP antibodies have also been associated with the development of airway disease [2]. There is growing evidence that rheumatoid arthritis begins in the lungs, a theory supported by a subgroup of patients who are anti-CCP positive with lung disease but have no articular manifestations [22, 23]. Additionally, a form of reactive lymphoid tissue known as inducible bronchus-associated lymphoid tissue (BALT) has been found in patients with rheumatoid arthritis-related lung disease and is associated with local production of inflammatory cytokines and anti-CCP antibodies [24]. A recent study examined the protein content in tissue samples obtained from lung and synovial biopsies of patients with rheumatoid arthritis, and found identical citrullinated vimentin peptides in both sites [18].

Schematic illustration of the concepts in the pathogenesis of rheumatoid arthritis associated-interstitial lung disease (RA-ILD). While the pathogenesis of RA-ILD is not fully understood, genetic predisposition and epigenetic factors are thought to play a role. Some patients with rheumatoid arthritis may have a genetic predisposition to ILD. Human leukocyte antigen (HLA)-B54, HLA-DQ1B*0601, HLA-B40, HLA-DR4 and the site encoding α-1 protease inhibitor have been associated with increased risk of ILD in rheumatoid arthritis, while some shared epitopes (HLA-DRB1) have been associated with the development of rheumatoid arthritis but decreased rates of RA-ILD. Male sex and older age are additional risk factors for RA-ILD. Both genetic factors and smoking are associated with increased citrullination of proteins in the lung, which allows for exposure of new epitopes and an autoimmune response. Higher levels of anti-cyclic citrullinated peptide (CCP) antibodies have been found in patients with RA-ILD compared to patients with rheumatoid arthritis only, but the role of such antibodies in the pathogenesis of RA-ILD is not clear. Smoking is also a risk factor for rheumatoid arthritis alone and results in repeated injuries to the alveolar epithelium and alterations in the cytokine milieu. As in other ILDs, the inflammatory response activates cytokines, chemokines and growth factors, such as tumor necrosis factor (TNF), vascular endothelial growth factor (VEGF), platelet derived growth factor (PDGF) and interleukins (IL). These contribute to a differentiation and proliferation of fibroblasts, increased synthesis and deposition of extracellular matrix (ECM) and increased activity of matrix metalloproteinases (MMP) resulting in ILD. Fibroblasts in the synovial lining play a similar role in the pathogenesis of joint manifestations of rheumatoid arthritis. (Figure prepared with assistance from Sean Mclaughlin; Seattle, WA, USA).
Cigarette smoking may play a role in inducing antibody formation and has been linked to higher titres of rheumatoid factor [20, 21]. Smoking may also play a specific role in RA-ILD by promoting citrullination of lung proteins, thus leading to the development of anti-CCP antibodies [25, 26]. This especially seems to be the case for individuals who have the shared epitope human leukocyte antigen (HLA)-DRB1. A large case–control study from Sweden demonstrated a 21-fold increased risk of developing rheumatoid arthritis among those who were anti-CCP positive, smoked and had two copies of the shared epitope gene versus nonsmokers who did not have the shared epitope gene [25]. It is thought that citrullination increases binding of peptides to HLA-DRB1 shared epitopes, therefore increasing immunogenicity of these proteins [25, 26]. A Japanese study evaluated the association between RA-ILD and specific HLA-DRB1 subtype alleles; while some alleles appeared to have a significant association, others appeared protective, and the majority had no significant association either way. This suggests that the shared epitope may have a role in the pathogenesis of rheumatoid arthritis, but not necessarily in the development of ILD [27].
In addition to de novo changes, medications used to treat articular manifestations of rheumatoid arthritis may play a role in development of ILD, which will be described later.
Pulmonary function tests
The majority of patients with RA-ILD will have a restrictive pattern on PFTs, with or without decreased diffusing capacity of the lung for carbon monoxide (DLCO) and hypoxaemia [7]. Impairment of both forced vital capacity (FVC) and DLCO is associated with poorer prognosis, but following systematic review of studies evaluating prognostic factors in RA-ILD, only DLCO had statistical significance after controlling for confounders [12]. Airflow obstruction may coexist and be seen in patients manifesting airway involvement, i.e. bronchiolitis obliterans.
Imaging
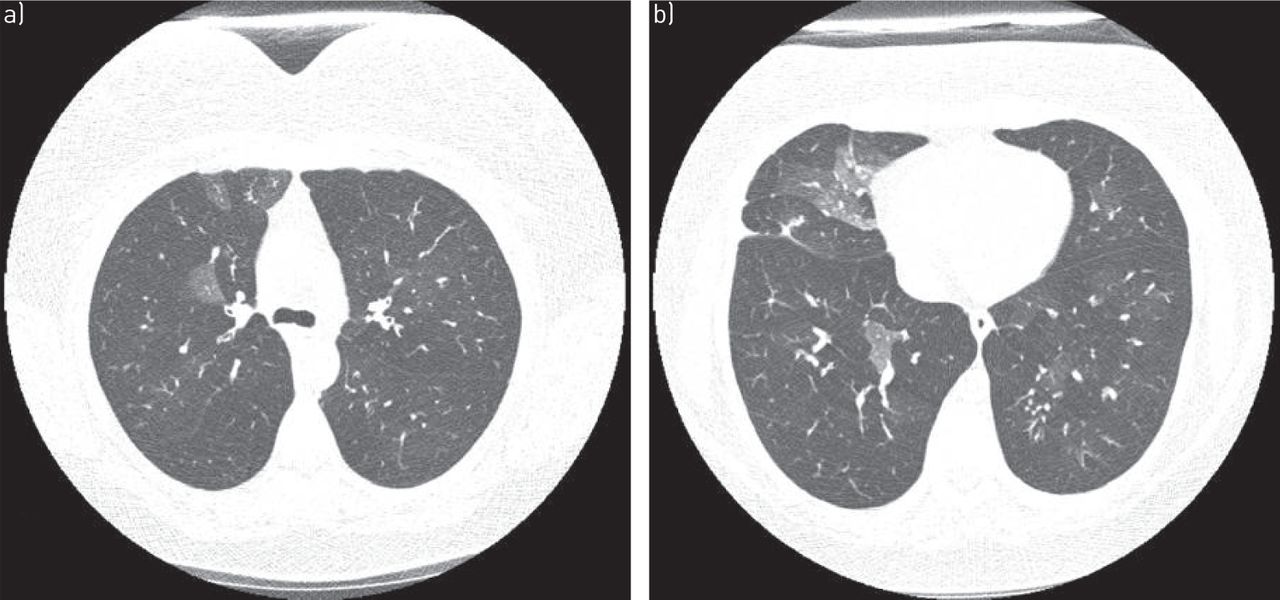
When considering a diagnosis of RA-ILD, knowledge of the histopathological pattern of ILD by surgical lung biopsy may provide information that assists in the diagnosis and prognostication, and has been considered the “gold standard” for diagnosis by some. While knowledge of the specific histopathological patterns of interstitial pneumonias may clarify histopathological diagnosis and be of prognostic value, this knowledge is usually not essential to determining a treatment regimen, which is invariably made up of immune modulating agents. Thus, in patients with known rheumatoid arthritis, and in the absence of clinical suspicion for infection and/or other respiratory complications, HRCT may be used to make a diagnosis of interstitial pneumonia. A variety of patterns are seen on HRCT scans in rheumatoid arthritis, with the most common being usual interstitial pneumonia (UIP), which occurs in 40–62% of cases [10, 28]. This is a notable difference from other connective tissue disorders, in which a nonspecific interstitial pneumonia (NSIP) pattern is most frequently seen [10, 29, 30]. In UIP, HRCT scans show subpleural, basal predominant, reticular abnormalities with honeycombing, and traction bronchiectasis but a relative absence of ground-glass opacities (fig. 2) [31]. Studies have shown there is good correlation between HRCT and surgical lung biopsy in patients with idiopathic pulmonary fibrosis (IPF), and surgical lung biopsy may not be necessary in patients with a HRCT scan demonstrating classic findings for UIP [2, 29, 32]. NSIP is the second most common pattern, occurring in ∼11–32% of patients [28]. NSIP is characterised by basilar predominant ground-glass opacities and absence of honeycombing (fig. 3). Additional patterns less commonly seen in rheumatoid arthritis include other patterns of interstitial pneumonia, including organising pneumonia, diffuse alveolar damage (DAD), lymphocytic interstitial pneumonia (LIP) and desquamative interstitial pneumonia (DIP)-like patterns. Combined pulmonary fibrosis and emphysema (CPFE) has also been demonstrated on HRCT scans in patients with rheumatoid arthritis. Patients typically have centrilobular or paraseptal emphysema in conjunction with lower lobe fibrosis, which is clinically relevant in that the CPFE pattern is associated with an increased risk of pulmonary hypertension [33, 34].

a) Axial and b) coronal computed tomography scans of usual interstitial pneumonia pattern in a patient with rheumatoid arthritis. Subpleural and basilar predominant reticulations, minimal ground-glass opacities, honeycombing (arrow) and pleural thickening (arrowhead) are visible, as well as traction bronchiectasis.
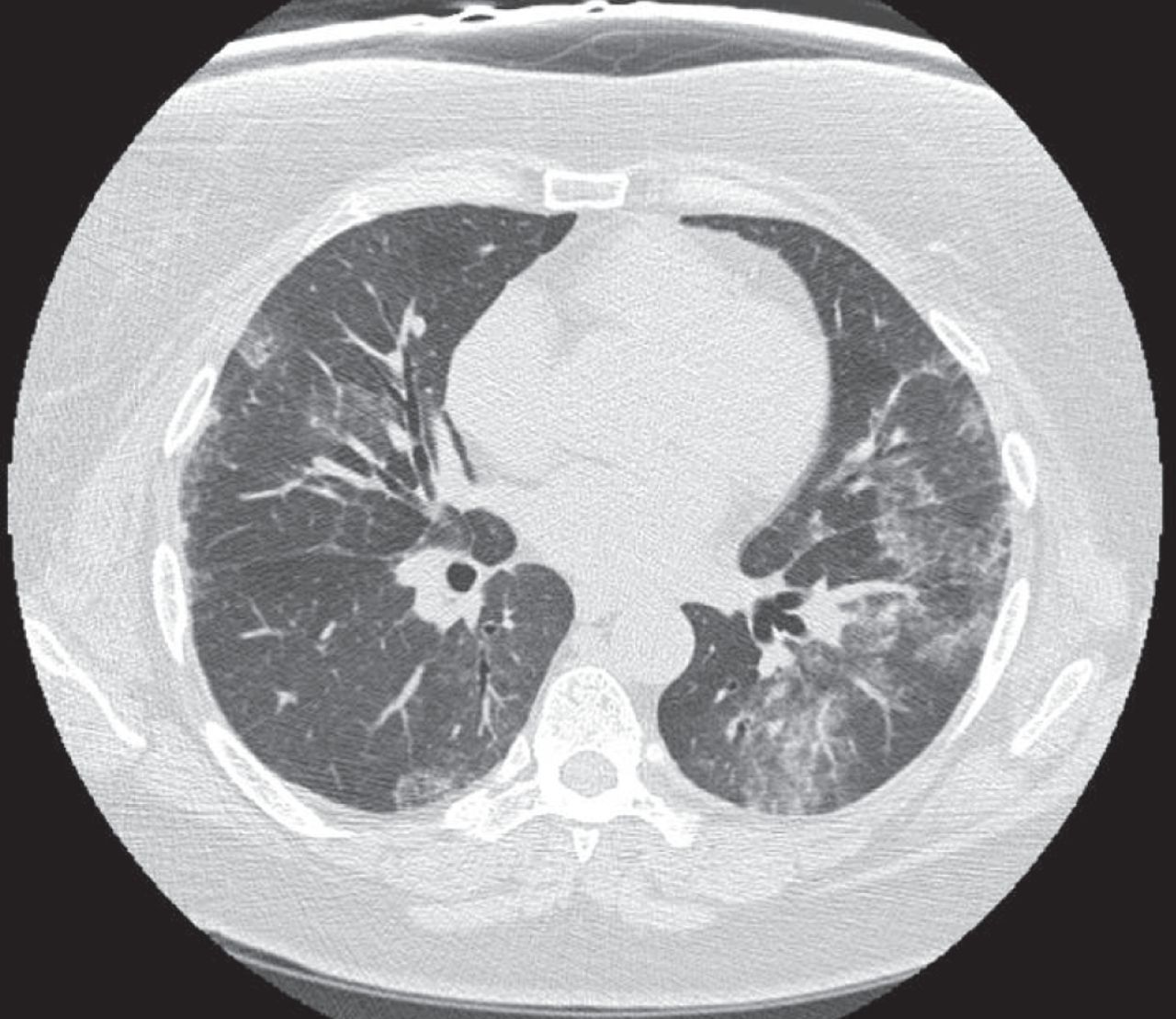
High-resolution computed tomography scan revealing a nonspecific interstitial pneumonia pattern with basal predominant ground-glass opacities and associated subpleural sparing in a patient with rheumatoid arthritis.
Pathology
Surgical lung biopsy varies depending on the underlying pattern, and may demonstrate UIP, NSIP, organising pneumonia, DIP, LIP or acute interstitial pneumonia. In UIP, a characteristic heterogenous pattern of fibroblast foci amid regions of normal tissue is seen. Subtle histopathological differences may be seen between rheumatoid arthritis-associated UIP and IPF-UIP; in rheumatoid arthritis-UIP there are often fewer fibroblast foci and a higher number of germinal centres [30, 35]. This may not be unique to the UIP pattern, as significantly higher numbers of CD4+ T-cells have been described in tissue samples from both UIP and NSIP when compared to purely idiopathic interstitial pneumonia [36]. Distinguishing among patterns of ILD can be useful in prognostication as patients with UIP or DAD have greater 5-year mortality compared to those with patterns such as NSIP or organising pneumonia [37].
Management
Treatment with anti-inflammatory and/or immunosuppressive agents is recommended regardless of the pattern of fibrosis. This is in contrast to IPF, in which use of immunosuppressive therapy has not demonstrated any clinical benefit. To date, there have been no randomised controlled trials comparing medications for the treatment of rheumatoid arthritis lung disease. Corticosteroids are the mainstay of therapy, particularly for cases of NSIP or organising pneumonia where they may lead to regression of consolidation on imaging and potential clinical improvement [28]. Cyclophosphamide and azathioprine have been used with varying success [38, 39], and there are a few case reports of ILD regression following cyclosporine treatment [40, 41]. More recently, several small studies have demonstrated stabilisation and/or improvement in symptoms, imaging and PFTs with use of mycophenolate [2, 42, 43]. A review of 125 patients with connective tissue disease associated-ILD (18 of which had rheumatoid arthritis) found that mycophenolate was associated with improvement in PFTs in patients with non-UIP patterns of ILD, and led to stabilisation among those with UIP [43].
Methotrexate, a first-line agent in the treatment of rheumatoid arthritis joint disease, is known to be associated with drug-induced pneumonitis, but fortunately this is rare. However, there is no evidence that this agent leads to progression of ILD. Following 6 weeks of treatment with high-dose steroids, one group found that treatment with methotrexate versus leflunamide or azathoprine was actually associated with an improvement in FVC at 6 months among patients with less fibrosis, although there was no evidence of differences in other outcomes such as mortality. This suggests that methotrexate use may not be associated with poorer outcomes than other disease modifying anti-rheumatic drugs [44]. There is considerable controversy as to whether anti-tumour necrosis factor (TNF) agents improve or worsen ILD. Studies evaluating this issue tend to be confounded by older age and prior use of methotrexate among participants. Similar controversy also exists for rituximab, with some studies reporting improvement [45] and other studies reporting development of ILD [46]. Risks and benefits of disease modifying anti-rheumatic drugs must therefore be weighed carefully, but in patients with significant pulmonary disease, potential benefits often outweigh risks of drug toxicity.
Adjuvant therapy for RA-ILD includes smoking cessation, management of gastro-oesophageal reflux disease, referral to pulmonary rehabilitation, supplemental oxygen, and vaccination against influenza and pneumococcal disease. In the absence of active rheumatoid arthritis, patients with rheumatoid arthritis lung disease who fail to respond to therapy should be considered for lung transplant. In patients with a UIP pattern, work-up for transplant should be considered early. A retrospective review of Canadian patients with advanced lung disease found no difference in outcomes between patients with RA-ILD and those with IPF at 1 year following lung transplant, suggesting that transplant is a reasonable option for these patients [47].
Prognosis
ILD is second only to cardiac disease as a cause of mortality in rheumatoid arthritis [1, 7, 10]. Based on a review of mortality data in the USA from 1988–2004, Olson et al. [3] calculated that ILD contributed to death in 6.8% of females and 9.8% of males with rheumatoid arthritis. Additional risk factors for mortality include advanced age, male sex, UIP pattern and extent of fibrosis on imaging or histopathology, and low DLCO [12]. Overall, RA-ILD has a better prognosis when compared to identical patterns in non-connective tissue disease-associated idiopathic interstitial pneumonias [48]. The possible exception to this is rheumatoid arthritis associated-UIP, which appears to have poorer prognosis compared to other patterns of RA-ILD and, in fact, may have similar outcomes to IPF [7, 29, 35, 49]. The mean survival for RA-ILD overall has been estimated at 2.6 years from time of diagnosis compared to 9.9 years for rheumatoid arthritis patients without lung involvement; however, this probably reflects the predominance of the UIP pattern [11].
Pleural disease
Pleural involvement is a common pulmonary manifestation of rheumatoid arthritis, with small pleural effusions noted in up to 70% on autopsy studies [50, 51]. However, only about 3–5% of patients are symptomatic [50, 51]. Pleural disease is more common in older (aged >35 years) males and those with rheumatoid nodules. Most effusions are unilateral, although occasionally bilateral effusions are found [50, 52]. Fever and pleuritic chest pain are common, but cough is generally absent unless there is comorbid parenchymal lung disease. Occasionally, a comorbid pericardial effusion may exist. Similarly, pneumothorax is possible, but rare.
Pathogenesis
A variety of mechanisms have been postulated for pleural effusions occurring in rheumatoid arthritis patients. These include impaired fluid resorption in inflamed pleura, necrosis of subpleural rheumatoid nodules, and local production of cytokines and immune complexes leading to endothelial injury and capillary permeability [50].
Imaging
Most cases of rheumatoid pleural disease can be diagnosed on chest radiography, with blunting of the costophrenic angles in the upright position. Fluid can also be detected on chest ultrasonograpy or computed tomography, with the latter being more useful if there is concern for comorbid parenchymal pathology (fig. 4). Computed tomography can also identify cavitating rheumatoid nodules, which can result in pneumothorax and/or bronchopleural fistula.

Computed tomography scans of a small unilateral pleural effusion (arrow) and pleural thickening in a patient with rheumatoid arthritis.
Pleural fluid studies
Thoracentesis should be performed for any effusion with >1 cm of layering on decubitus films. The typical “rheumatoid effusion” is a sterile exudative fluid with low pH (<7.3), low glucose (<60 mg·dL−1) and elevated lactate dehydrogenase (may be >700 IU·L−1) [50, 51, 53]. Low pH results from elevated glucose metabolism in the inflamed pleural space, with resultant production of lactate and carbon dioxide [50, 52]. Fluid glucose levels may be similar to serum glucose levels in acute disease, but typically fall quite low in chronic effusions. It is speculated that this may be due to pleural thickening reducing the ability of glucose to cross into the pleural space, or due to consumption from inflamed pleura [50]. Chronic pleural inflammation results in the presence of cholesterol crystals in the fluid, resulting in a milky appearing “pseudochylous” pleural fluid. This is in contrast to true chylothorax occurring from lymphatic rupture, where triglycerides and/or chylomicrons are found in the fluid. Rheumatoid factor is often present in pleural fluid, and may be higher than serum levels.
White cell count and cell predominance is variable. Characteristic elongated multinucleated macrophages, “ragocytes” (polymorphonuclear phagocytes with intracellular inclusions of IgG and/or rheumatoid factor), or necrotic background debris may be seen. An analysis of 29 cases of rheumatoid pleural effusion for which fluid studies were reported noted predominance of neutrophils, lymphocytes and eosinophils in 56%, 37% and 15% of cases, respectively. In patients who had multiple thoracenteses, a transition from neutrophil-predominant to lymphocyte-predominant fluid was noted over a 7–11-day period. Interestingly, all patients with eosinophil-predominant fluid lacked a preceding diagnosis of rheumatoid arthritis; this was either diagnosed concurrently or at a later time [54].
Infection should always be ruled out, particularly as the low pH, low glucose and high lactate dehydrogenase seen in rheumatoid effusions is also typical for empyema. Sterile “empyematous” effusions may be the result of a ruptured necrotic subpleural rheumatoid nodule into the pleural space and subsequent bronchopleural fistula. Longstanding pleural inflammation can result in the formation of a fibrous peel, resulting in a trapped lung, where the lung is unable to re-expand after drainage of pleural effusion.
Tuberculosis and certain malignancies can mimic rheumatoid effusions. Although not necessary for the diagnosis of rheumatoid pleural effusion, video-assisted thorascopy with pleural biopsy is undertaken when the diagnosis is unclear. In rheumatoid pleuritis, the parietal pleural is thickened with a “gritty” granular appearance. On histology, there is replacement of the normal mesothelial lining with multinucleated giant cells and foci of palisading fibroblasts and lymphocytes surrounding necrotic centres, similar to rheumatoid nodules [52, 53].
Management
Most cases of rheumatoid pleuritis improve with treatment of the underlying rheumatoid arthritis; effusions that are small and asymptomatic do not require specific intervention [50]. In a case series involving nine patients with rheumatoid arthritis and pleural effusions, all had resolution of the effusion within 3 years, with an average time to resolution of 14 months. No specific treatment was used other than therapeutic thoracentesis when indicated [53]. An earlier case series reported resolution by 3 months in 13 out of 19 patients; only two patients received corticosteroids. However, in this same series, one patient had a persistent effusion that resulted in severe pleural thickening and trapped lung, which ultimately required decortication [55]. This would argue that patients with large or persistent effusions should be treated to avoid similar complications.
Airway disease
Conditions affecting both the upper and lower airways can occur in patients with rheumatoid arthritis.
Upper airway involvement
Upper airway disease occurs more frequently in females and those with longstanding or severe disease [56, 57]. Manifestations include rheumatoid nodules on the vocal cords, vasculitis affecting the recurrent laryngeal or vagus nerves leading to vocal cord paralysis, or arthritis of the cricoarytenoid joint. In the latter condition, synovial thickening and build-up of excess synovial fluid leads to progressive cartilage erosion and subluxation of the joint. These findings are best seen on HRCT scans of the neck and are often present before clinical symptoms develop [58, 59]. Patients may have symptoms of dysphagia, throat pain or fullness, or exertional dyspnoea, but many are asymptomatic until significant obstruction occurs [60]. Acute stridor or obstructive respiratory failure may occur from sudden subluxation or superimposed airway oedema from infection or intubation. Mild symptoms may be managed with nonsteroidal anti-inflammatory drugs or rheumatoid arthritis-directed therapy. For more severe obstruction, surgical intervention may be required in addition to immediate airway management [56, 60].
Lower airway involvement
Lower airway disease may include bronchial hyperresponsiveness, bronchiolitis or bronchiectasis. Similar to RA-ILD, estimates of the prevalence of obstructive airway disease are highly variable depending on the criteria used to define disease and the population studied [61]. In addition, studies attempting to correlate small airways disease with rheumatoid arthritis are often confounded by smoking or the presence of other RA-ILD. A small prospective study of 50 patients with rheumatoid arthritis and no known ILD found that 70% had evidence of lower airway disease on HRCT; however, 20% of these patients were smokers [62]. A retrospective cohort study from Taiwan found a higher rate of chronic obstructive pulmonary disease (COPD) in patients with rheumatoid arthritis compared to those without, particularly in young adults aged 20–34 years [63]. However, the authors were unable to assess differences in smoking habits between study groups. A longitudinal study evaluating asymptomatic nonsmoking patients with rheumatoid arthritis found a slightly higher rate of PFT abnormalities at baseline (8.7% versus 5% of the reference population), but this number did not significantly change over the course of 10 years, leading the authors to question the significance of PFT abnormalities in patients without respiratory symptoms [64].
Follicular bronchiolitis occurs in the setting of hyperplasia of BALT and may be seen in a variety of connective tissue disorders, including rheumatoid arthritis. HRCT demonstrates centrilobular peribronchial nodules <3 mm in size with branching structures corresponding to bronchial dilation and wall thickening; honeycombing is absent. Pathology shows hyperplastic lymphoid follicles with germinal centres adjacent to airways [65, 66]. PFTs usually demonstrate a restrictive pattern, although obstruction may be noted as well. Treatment is directed at the underlying rheumatoid arthritis, and additional treatment may not be necessary for mild disease. For more severe or symptomatic disease, corticosteroids and macrolide antibiotics have been used [65].
Obliterative bronchiolitis (also referred to as constrictive bronchiolitis) is a more severe and often fatal condition characterised by progressive narrowing of the bronchioles. It is more common in females and those with positive rheumatoid factor and longstanding untreated disease, and may also occur in the setting of certain medications including gold, penicillamine and sulfasalazine. In contrast to other rheumatoid lung manifestations, obliterative bronchiolitis presents acutely with rapidly progressive dyspnoea, cough and bronchorrhea in the absence of other systemic symptoms. HRCT findings are nonspecific, but may show centrilobular emphysema, bronchiectasis, bronchial wall thickening or mosaic attenuation (fig. 5). PFTs generally show airflow obstruction with a normal DLCO. The mainstay of treatment is to discontinue the offending agent, which will occasionally result in the regression of symptoms. However, the overall prognosis is poor [67]. High-dose corticosteroids are often used, although they rarely have an impact [68]. Azathioprine and cyclophosphamide have been used, although it is unclear whether these agents have any efficacy [68, 69]. A few case reports have described some improvement with anti-TNF therapy [70]. Macrolide antibiotics, in particular erythromycin, may also be effective [65, 68]. In severe cases, lung transplant may be necessary.

a, b) Expiratory computed tomography scans of constrictive bronchiolitis with areas of mosaic attenuation consistent with air trapping in a patient with rheumatoid arthritis.
Bronchiectasis has been demonstrated on HRCT in ∼30% of cases of rheumatoid arthritis, although it may be clinically silent [71, 72]. Bronchiectasis may precede or follow the development of rheumatoid arthritis [73]. Various hypotheses exist regarding the association between bronchietasis and rheumatoid arthritis, including: chronic suppurative infections leading to bronchiectasis, which is perhaps enhanced in the setting of rheumatoid arthritis; or treatment with disease modifying anti-rheumatic drugs, or alternatively that chronic infections in a bronchiectasis patient provide additional antigenic stimuli that then triggers rheumatoid arthritis [72, 73]. It is also hypothesised that rheumatoid arthritis and bronchiectasis share a genetic predisposition [73]. A French study found that patients with rheumatoid arthritis and symptomatic bronchiectasis were more likely to be heterozygous for the ΔF508 mutation, compared to those with rheumatoid arthritis without bronchiectasis and those with bronchiectasis of unknown aetiology [74]. When bronchiectasis is severe enough to produce clinical symptoms, it may complicate the use of immunosuppressive medications, particularly anti-TNF agents as both bronchiectasis and anti-TNF therapy increase the risk of certain pulmonary infections. Among patients with rheumatoid arthritis and bronchiectasis, mortality rates are higher than for either condition alone [72]. There are no specific guidelines for the management of rheumatoid arthritis with bronchiectasis, and therapy is the same as for either condition alone, with bronchodilators, antibiotics and bronchial hygiene used to treat bronchiectasis.
Pulmonary nodules
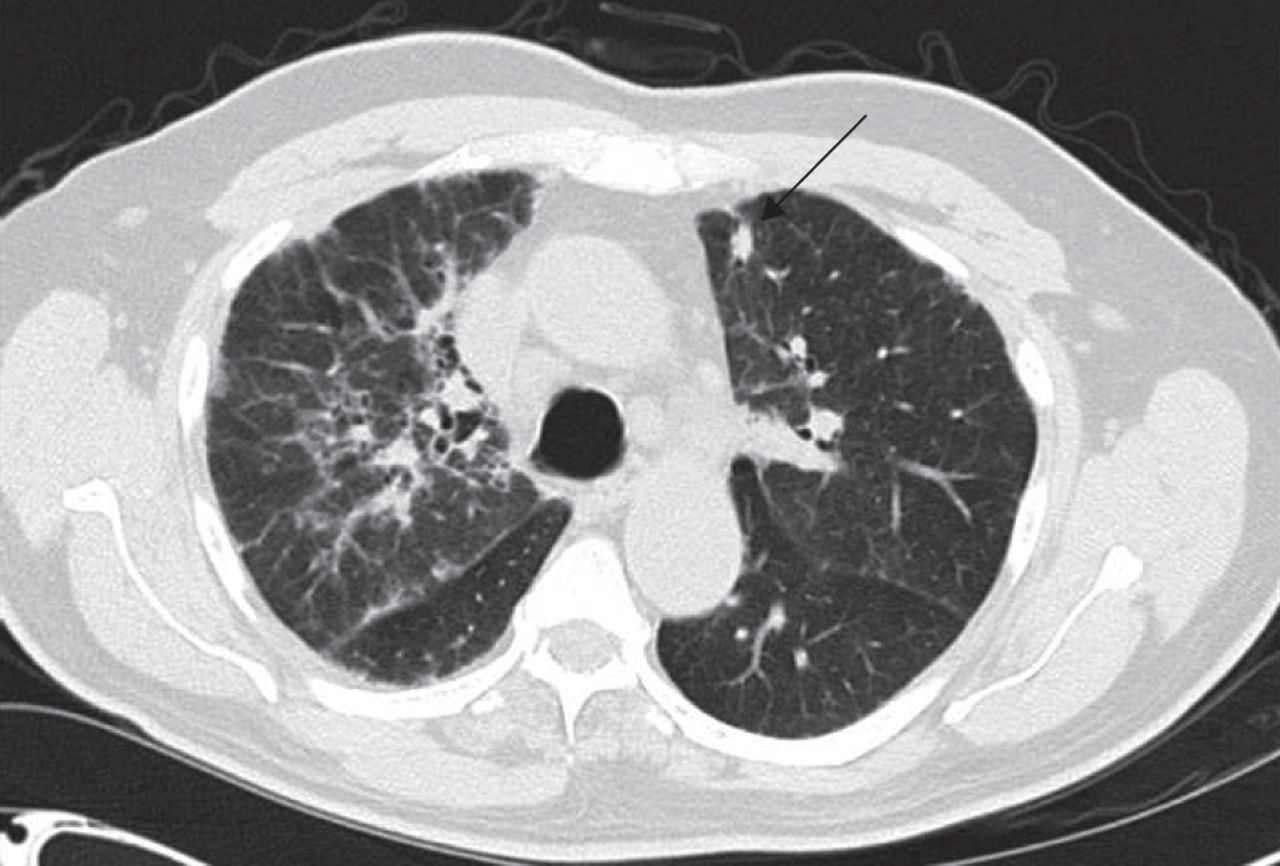
Rheumatoid nodules can occur in the lungs, particularly in patients with longstanding disease and subcutaneous nodules. They are typically located along the interlobular septa or in subpleural regions. Nodules may be single or multiple, ranging in size from a few millimetres to several centimetres (fig. 6). Pathological examination shows central fibrinoid necrosis with palisading mononuclear cells and associated vasculitis [75]. Nodules are typically asymptomatic unless they cavitate or rupture, in which case infection, pleural effusion or bronchopleural fistula may occur. Uncomplicated nodules may spontaneously regress or improve with standard rheumatoid arthritis therapy. However, rheumatoid nodules have, at times, been noted to paradoxically enlarge with rheumatoid arthritis treatment, in particular, this has been observed with methotrexate treatment [76]. In patients who are past or current smokers, it is important to differentiate nodules from malignancy. Prior imaging studies and Fleischner Society Guidelines may be used to guide further evaluation of solitary pulmonary nodules [77]. Positron emission tomography scans may be used in the evaluation of nodules ≥8 mm in diameter; in general, rheumatoid nodules show little or no uptake on positron emission tomography scans, although increased uptake may be seen if active inflammation is present [78].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Computed tomography scan of solitary subpleural pulmonary nodule (arrow) in a patient with rheumatoid arthritis.
A rare complication known as Caplan syndrome (also known as rheumatoid pneumoconiosis) may occur in those with pneumoconiosis from occupational exposure to coal, silica or asbestos. This is characterised by sudden development of multiple peripheral pulmonary nodules. These lesions may coalesce and cavitate after a period of rapid growth over weeks to months; they typically remain unchanged for years. Classically patients are rheumatoid factor positive and have mild exposure pneumoconiosis at baseline; however, patients may develop nodules in the absence of pre-existing joint or lung disease [79]. Pathologically, nodules are similar to other rheumatoid nodules but typically have rings of dust surrounding and within an area of central necrosis. This region is surrounded by a zone of cellular infiltration consisting of granulocytes and macrophages (which may contain dust particles). Patients with this syndrome are often asymptomatic and the overall prognosis is good. Complications occur when a lesion cavitates and becomes infected or ruptures into the pleural space [79].
Vascular disease
Pulmonary hypertension can occur in rheumatoid arthritis-associated lung disease, usually in the setting of parenchymal lung involvement. However, isolated pulmonary hypertension has also been described [80–83]. Udayakumar et al. [81] compared 45 patients with rheumatoid arthritis to 45 healthy age-matched controls, and found a significantly higher rate of asymptomatic pulmonary hypertension (defined as pulmonary artery systolic pressure ≥30 mmHg by Doppler echocardiography) among those with rheumatoid arthritis (26.7% versus 4.5%). This was especially true for older patients and those with longer disease duration. Patients with traditional risk factors for cardiopulmonary disease were excluded from the study, and only three (6.7%) patients with rheumatoid arthritis had coexisting clinically significant fibrotic lung disease. This meant 20% of the rheumatoid arthritis patients included in this study had isolated pulmonary hypertension by echocardiography [81]. A similar study evaluating 40 Turkish patients with rheumatoid arthritis found that 11 (27.5%) had pulmonary artery systolic pressure ≥30 mmHg on echocardiography; presence of joint deformity was the only significant difference between those with pulmonary hypertension and those without [82]. These studies are in agreement with an earlier, larger study involving 146 patients with RA, in which 21% of patients were found to have mild-to-moderate pulmonary hypertension on echocardiography in the absence of clinically significant heart or lung disease [83]. In each of these studies none of the patients found to have pulmonary hypertension were symptomatic. It is possible that these patients are less active as a result of their arthritis, and perhaps do not notice dyspnoea until they have more advanced cardiopulmonary disease. This raises the question of whether patients with rheumatoid arthritis should undergo regular screening for pulmonary hypertension, particularly those with longer standing disease, although there are currently no guideline recommendations for such screening. Patients who manifest pulmonary hypertension may benefit from the use of medications indicated for the treatment of pulmonary hypertension associated with connective tissue disease.
Patients with rheumatoid arthritis are also at increased risk of venous thromboembolism, both deep venous thrombosis and pulmonary thromboembolism, compared to those without RA, even after adjusting for age, sex and comorbid diseases [84, 85]. Patients with rheumatoid arthritis and more severe extra-articular disease are at even greater risk of venothromboembolism, supporting the hypothesis that some of the increase in risk is attributable to prothrombotic effects of chronic inflammation [86, 87].
Drug toxicity
Most patients with diagnosed rheumatoid arthritis are on disease-modifying or immunosuppressant therapy to treat the joint manifestations. Theoretically, these medications should protect the lungs by reducing levels of inflammatory cytokines, which are known to be elevated in some patients with rheumatoid arthritis [88]. Several of these medications have been implicated in the development of lung disease, although it is often difficult to prove causality as patients with rheumatoid arthritis are prone to lung complications from infection, other medications and the disease itself. In addition, ILD that progresses while on a particular therapy may represent treatment failure rather than an effect of the implicated medication.
Methotrexate
Methotrexate is the most common first-line agent used to treat rheumatoid arthritis that prevents joint destruction. A possible link between this medication and lung disease was first reported in 1983; since then many more cases have been reported [89]. Acute/subacute hypersensitivity pneumonitis has been well-described in the literature, with a variable incidence ranging from 0.86% to 6.9% in treated patients, with higher dose methotrexate more likely to be associated with pulmonary toxicity [90]. This typically occurs within the first year of treatment and is felt to represent a hypersensitivity reaction [7, 91]. Symptoms include dyspnoea and nonproductive cough with/without systemic symptoms. Imaging findings are relatively nonspecific, with diffuse pulmonary opacities or patchy consolidation seen on chest radiographs and HRCT. Chest radiographs may be normal in the early stages of disease. BAL and lung biopsy are more helpful in ruling out alternative causes (i.e. infection) than in establishing the diagnosis of methotrexate-induced lung injury, although the presence of poorly formed non-necrotising granulomas and scattered eosinophils may suggest methotrexate-induced hypersensitivity pneumonitis, as these are not typical findings in RA-ILD [89, 91, 92]. Therapy consists of stopping the medication; most patients will have clinical improvement within days with radiological improvement over the course of several weeks. In more refractory cases, glucocorticoids may be used. Rechallenging with methotrexate after recovery is generally not recommended; one study reported a recurrence rate of pneumonitis of ∼25% [92].
A more chronic, progressive pulmonary fibrosis has been described in the setting of methotrxate treatment, but it is controversial whether this is directly related to methotrexate. A study of 128 patients with rheumatoid arthritis compared patients treated with methotrexate to patients who had never received the drug and reported similar rates of pulmonary fibrosis on HRCT between the two groups and no difference in PFT decline when these groups were followed over 2 years [93]. Rarely, patients on methotrexate can develop an acute, severe, life-threatening pneumonitis characterised by respiratory failure and a DAD pattern on histopathology; however, this is difficult to differentiate from acute DAD arising from rheumatoid arthritis alone [10]. Notably, periodic pulmonary function testing has not been associated with the ability to detect methotrexate-associated pneumonitis prior to the development of symptoms, and serial PFT testing for this purpose is not routinely recommended [94]. In addition, methotrexate is known to provoke rheumatoid nodule formation. There have been reports of lymphoproliferative diseases developing in the setting of methotrexate treatment, with disease regression once medication is stopped. A recent meta-analysis of 22 studies involving rheumatoid arthritis patients treated with methotrexate (n=4544) versus other agents (including disease modifying anti-rheumatic drugs and biological agents) found a small increase in risk of respiratory infections (RR 1.11, 95% CI 1.02–1.21), but not in noninfectious complications, such as pneumonitis, or death from pulmonary causes among those treated with methotrexate [95].
Risk factors for developing lung disease in the setting of methotrexate use are not well known. A small case–control study from Australia found that patients who developed pneumonitis were more likely to have had pre-existing lung disease and shorter duration of therapy, although neither trend reached statistical significance [96]. A subsequent larger, multicentre, case–control study found an association between increasing age, previous treatment with other disease-modifying anti-rheumatic drugs (particularly gold, sulfasalazine and D-penicillamine), extra-articular manifestations, presence of diabetes and hypoalbuminemia with the development of methotrexate-associated pulmonary disease. The investigators also noted the risk was inversely related to the length of therapy, with most cases of pneumonitis occurring within the first 32 weeks of therapy [97]. Genetic predisposition to drug sensitivity may play a role as well. A recent case–control study of Japanese patients with rheumatoid arthritis who were treated with methotrexate found a significant association between the development of pneumonitis and the presence of the HLA-A*31:01 allele [98]. Cigarette smoking has not been shown to be a risk factor for the development of methotrexate-associated pulmonary toxicity.
Leflunomide
Leflunomide is typically used as second-line therapy after a patient has failed, or has contraindications, to methotrexate. It has been associated with the development and/or exacerbation of ILD, potentially secondary to an active metabolite that may induce transition of lung epithelial cells to myofibroblasts, a process known as the epithelial-mesenchymal transition [99]. However, in an animal model, administration of leflunomide alone did not result in this phenomenon. Rather, the process was enhanced when leflunomide was administered in the setting of bleomycin, a known profibrotic agent [99]. This suggests that leflunomide could provoke the development of ILD in predisposed populations. Interestingly, rates of leflunomide-related ILD appear to be higher in Asian populations (∼1%) compared to Western countries (<0.1%), again suggesting a potential genetic predisposition to drug sensitivity [89]. It is important to note that in at least one of these studies, all patients treated with leflunomide had been previously exposed to methotrexate, which may have been a confounding factor [100].
TNF-α inhibitors
Like methotrexate and leflunomide, TNF-α inhibitors have been associated with the development of ILD, but clear causality has been difficult to prove. Reports of new-onset ILD have been described for all five TNF-α inhibitors currently approved for the treatment of rheumatoid arthritis [7, 46, 101, 102]. Studies have reported sarcoid-like granulomatous disease, organising pneumonia and exacerbation of pre-existing ILD; however, most of these studies have been small and infection was not clearly ruled out [10, 28, 89]. One study assessed 122 cases of new-onset or exacerbated ILD in the setting of TNF-α inhibitor use, 108 cases were patients with rheumatoid arthritis. Of note, 63% of these patients had been treated with methotrexate, and 38% had pre-existing ILD. 15 (29%) patients who died were aged >65 years and had prior ILD, with longer duration of ILD being associated with risk of death [46]. In contrast, a large cohort study of 8417 patients with autoimmune disease did not show any significant difference in the incidence of ILD between those who were treated with anti-TNF therapy (0.5%) and those were treated with other therapies (0.3%). The incidence of ILD occurring in patients with rheumatoid arthritis was seven times higher than that for other connective tissue diseases, but again no significant difference was noted between those who received anti-TNF therapy and those who did not [103]. A variety of mechanisms for TNF-α inhibitor-induced ILD have been proposed, but no definite aetiology has been established.
Rituximab
Rituximab was originally used for the treatment of lymphoma, and the majority of safety data comes from the study of cancer patients. Pulmonary toxicity has rarely been reported in such patients treated with rituximab, and is calculated to occur in <0.03% of 540 000 treated cases [104]. Rituximab is now also used for the treatment of rheumatoid arthritis. However, the majority of respiratory adverse events continue to occur in those patients with haematological conditions and probably represent a tumour response [88, 105]. There has been scattered case reports of organising pneumonia associated with rituximab in rheumatoid arthritis, which improved with prednisone therapy [106]. However, a randomised controlled trial evaluating the efficacy and safety of rituximab in 465 patients with rheumatoid arthritis did not note any correlation with ILD [107]. In fact, small case studies have suggested a beneficial effect of rituximab on connective tissue disease-associated ILD, with one retrospective review reporting improvement or stabilisation of PFTs in 28 out of 33 patients with severe ILD [45]. The need for prospective, randomised clinical trials for RA-ILD/pulmonary fibrosis is evident.
Other medications
Other agents used in the treatment of rheumatoid arthritis have been implicated in lung disease. These include: nonsteroidal anti-inflammatory drugs and, the now rarely used, gold, which causes organising pneumonia; sulfasalazine and penicillamine, which have been associated with obliterative bronchiolitis; and azathioprine and tacrolimus, which have been reported to exacerbate pre-existing ILD [28]. Gold, an agent which is currently rarely used, was associated with lung toxicity in ∼1% of treated patients [10]; only about one-third of the cases were responsive to corticosteroids [108]. Sulfasalazine can also cause eosinophilic pneumonia, which typically improves with drug cessation [10]. There have been a few reports of noninfectious pneumonia developing in the setting of the anti-IL6 agent tocilizumab [101, 109, 110], as well as reports of exacerbations of pre-existing ILD [111, 112]. Abatacept, an inhibitor of T-cell co-stimulation that binds B7 (CD80 and CD86) on antigen presenting cells, has been associated with COPD exacerbations, but there has only been one report of possible ILD exacerbation described in the literature [113]. However, as for the other agents discussed, it is difficult to prove causality, as use of other agents, infection and rheumatoid arthritis itself can all lead to the development of ILD. Patients manifesting pulmonary hypertension will benefit from treatment of pharmacological agents indicated for pulmonary hypertension in the setting of connective tissue disease [114].
Infectious complications in patients with RA-ILD treated with immune modulating agents are, in general, opportunistic lung infections. When patients manifest new ground-glass changes superimposed on their baseline underlying ILD, this is always a concern and in the appropriate clinical setting patients must be evaluated with appropriate diagnostic interventions. It is difficult to understand the exact prevalence of lung infections related to rheumatoid arthritis medications as rheumatoid arthritis alone is known to be a predisposing risk factor for infection [115]. Several medications used to treat rheumatoid arthritis have been implicated with increased risk of lung infections; much of the literature involves glucocorticoids and anti-TNF agents [116–121]. As glucocorticoids are effective immunosuppressive medications, their use has been related to increased risks of lower respiratory tract infections, including influenza [116, 117]. A recent meta-analysis of observational studies noted a dose-dependent, increased risk of serious infections in patients with rheumatoid arthritis being treated with glucocorticoids [119]. TNF-α is involved in the host defence against invasions of viruses and bacteria. Particularly, mycobacterial disease has been highly associated with anti-TNF therapy [118, 120, 121]. A retrospective study found a high incidence of active tuberculosis soon after starting therapy with infliximab (median time 12 weeks) leading to the recommendation that patients started on anti-TNF therapy should be screened for latent tuberculosis infection [121]. Pneumocystis jiroveci pneumonia prophylaxis is often considered in patients taking immunomodulating medications. Although there are no published guidelines regarding Pneumocystis jiroveci pneumonia prophylaxis in patients with rheumatoid arthritis, our practise is to place patients on Pneumocystis jiroveci pneumonia prophylaxis if they are taking a dose of prednisone equivalent to 20 mg daily or higher in combination with another immunomodulating medication. Other noninfectious, complications of treatment with azathioprine include increased risk of lymphoma and malignant disorders [122, 123].
Conclusion
Pulmonary involvement is common among patients with rheumatoid arthritis and has a variety of manifestations, with ILD, pleural disease and pulmonary drug toxicity being the most common. Mechanisms of lung injury have been attributed to genetics, environmental exposure and medications. Pulmonary disease may precede the development of other rheumatoid arthritis manifestations, such as articular involvement, but patients with pulmonary disease may also be asymptomatic. Overall, morbidity and mortality from rheumatoid arthritis associated-lung disease are high. To date, there are no prospective randomised clinical trials for RA-ILD and, thus, treatment for RA-ILD is essentially immunomodulating agents that are used for rheumatoid arthritis in general. Further research is needed to determine specific risk factors and appropriate therapy.
Footnotes
Conflict of interest: None declared.
Provenance: Submitted article, peer reviewed.
- Received September 2, 2014.
- Accepted November 21, 2014.
- Copyright ©ERS 2015.
ERR articles are open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References









