





In recent years, there have been important advances in the understanding of diffuse parenchymal lung diseases, especially in relation to the idiopathic interstitial pneumonias, including idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia. Among other diffuse parenchymal lung diseases, the topics of particular interest centred on hypersensitivity pneumonia, parenchymal disease associated with connective tissue disease, lymphangioleiomyomatosis and pulmonary hypertension. Insights on disease prevalence, diagnosis, prognostic evaluation, and management for the granulomatous diseases have not been discussed in this review.
IDIOPATHIC PULMONARY FIBROSIS
Epidemiology
Idiopathic pulmonary fibrosis (IPF) is a devastating disease, with a 5-yr survival rate of only 20–30% following diagnosis. Pulmonary fibrosis remains a significant health problem whose true incidence and prevalence has been believed to be increasing in recent years. Using a database from the National Center for Health Statistics, Olson et al. [1] clearly show for the first time that the mortality rates have increased from 1992 to 2003. Although these data do not confirm that the prevalence is increasing, they do suggest that there have been improvements in reporting and identification of fibrotic lung diseases. The age-adjusted mortality increased for both sexes, and the authors of this report suggest that this trend will continue. The most common cause of death in people with IPF is progression of their underlying lung disease, but there is evidence that the incidence of other diseases, such as lung cancer, may be increased in people with IPF [2–5]. This is interesting, because such associations may shed light on the aetiology and/or pathogenesis of IPF, and highlight the need for a comprehensive approach to the care of people with IPF [6]. A previous review by Panos et al. [2] highlighted a possible increase in cardiovascular disease in people with IPF, and evidence supporting this observation comes from a recent uncontrolled autopsy study in which nine out of 42 people with IPF died from a cardiovascular event [4]. Furthermore, in a study of 630 people referred for lung transplantation, Kizer et al. [7] found that people with IPF were more than twice as likely to have angiographic evidence of coronary artery disease as people with nonfibrotic lung diseases. Findings from a recent study suggest that patients with IPF have a marked relative increase in the risk of vascular disease (angina, acute coronary syndromes, and deep-vein thrombosis) [8], and this should be considered during the routine care of these patients.
Prognostic evaluation in IPF
The clinical course of patients with IPF is variable and can display long periods of stability, a steady gradual decline, and/or periods of acute deterioration [3, 9]. This unpredictable nature of usual interstitial pneumonia (UIP)/IPF highlights the importance of identifying factors that can help refine the prognosis for patients at the time of initial diagnosis. One of the most striking findings of a recent study [10] is the variable high-resolution computed tomography (HRCT) appearance of UIP despite very rigid histopathological criteria. It would have been expected that the rigid inclusion criteria define a more homogeneous patient population with similar HRCT features. Interestingly, only approximately one-third of HRCTs showed definite IPF (figs 1⇓ and 2⇓) and approximately one-third suggested an alternative diagnosis, such as nonspecific interstitial pneumonia (NSIP) (fig. 3⇓), or were unclassifiable. This study highlights the critical importance of a surgical lung biopsy in making an accurate diagnosis in patients without definite HRCT appearance of UIP, even when an alternative diagnosis, such as NSIP, is suspected [11]. This study also sheds additional light on the role of HRCT in determining prognosis for patients with UIP/IPF. The survival of patients within each HRCT category was statistically similar, although the median survival was shortest for the definite UIP group (35 months), intermediate for the consistent with UIP group (43 months), and longest for the alternative diagnosis group (112 months). The lack of statistical significance in the study could be due to limited sample size or selection bias [10].

Definite usual interstitial pneumonia pattern. High-resolution computed tomography demonstrates honeycombing in a predominantly peripheral distribution.

A pattern consistent with usual interstitial pneumonia pattern. High-resolution computed tomography image demonstrates irregular intralobular reticular opacity, ground-glass opacity and traction bronchiectasis in a predominantly peripheral distribution. There is no extensive honeycombing.
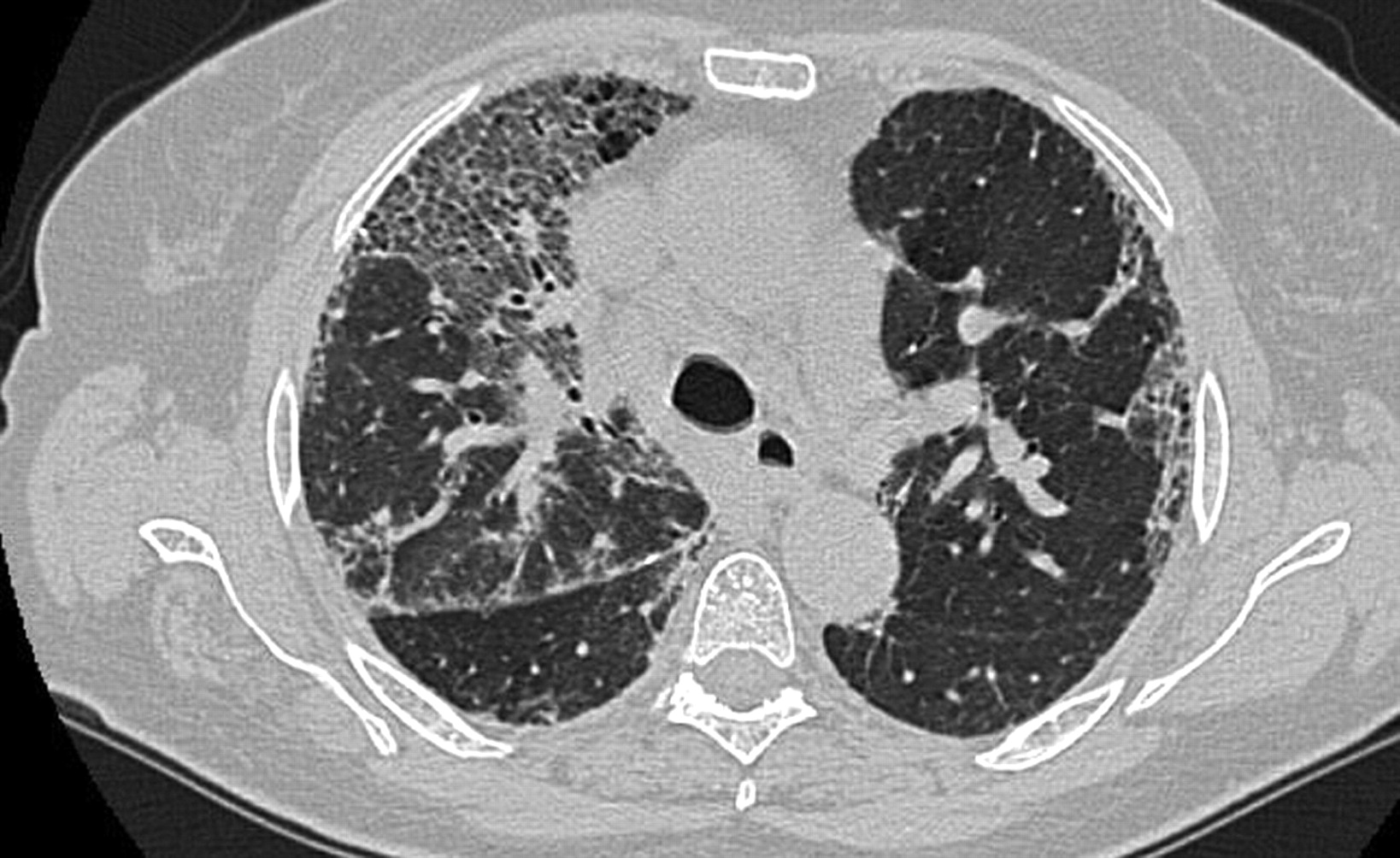
Pattern high-resolution computed tomography consistent with nonspecific interstitial pneumonia in a patient with histological diagnosis of usual interstitial pneumonia. Ground-glass opacities, reticulation and marked traction bronchiectasis are present in the lung.
A decrease in forced vital capacity (FVC) of at least 10% or diffusing capacity for carbon monoxide (DL,CO) of at least 15% over 6 or 12 months is associated with decreased survival [12, 13]. Although change in FVC is a good surrogate for subsequent mortality, it is imperfect as some patients die without a 10% decline in FVC, whereas others can live for prolonged periods even after a 10% decline in FVC [3, 14]. Distance walked during 6-min walk test (6MWT) was an important prognostic factor in recent studies [15, 16]; however, the prognostic value of desaturation and distance walked during 6MWT remain controversial. Maximal oxygen uptake (V′O2,max) is an integrated measure of cardiovascular, respiratory, and neuromuscular function. In a recent study, a baseline V′O2,max <8.3 mL·kg−1·min−1 threshold was identified in IPF patients, below which the risk of death was greatly increased [17].
An area of debate is the utility of bronchoalveolar lavage (BAL) for diagnosis and/or prognosis in IPF. Although the pattern of cells recovered with this procedure may narrow the differential diagnosis of interstitial pneumonias, its role in IPF remains controversial. A previous report suggested that BAL lymphocytosis >20% is associated with response to anti-inflammatory therapy and improved survival in biopsy-proven patients with IPF [18]. Kinder et al.[19] report that a low percentage of neutrophils (<3%) is associated with favourable outcomes, and that each doubling of baseline BAL fluid neutrophil percentage is associated with a 30% increased risk of mortality during the first year after diagnosis. The presence of BAL lymphocytosis was shown to shift diagnostic probabilities from IPF to hypersensitivity pneumonitis or sarcoidosis. In patients with IPF, as then diagnosed, BAL lymphocytosis denoted a better outcome from treatment. These data suggest that the utility of BAL for risk stratification of patients with IPF should be reconsidered [20, 21]. There may be a complementary role for BAL with cellular differentials in diagnosing interstitial lung diseases (ILDs) in a minority of subjects. In patients suspected of IPF clinically, without classic computed tomography features for IPF (absence of honeycombing or a substantial amount of ground-glass opacification outside of areas of reticulation) and who are at high risk for surgical lung biopsy, BAL may be a reasonable tool to use. Surfactant protein (SP)-A and SP-D are members of the collectin family. Secreted primarily by alveolar epithelial type II pneumocytes, plasma SP-A and SP-D levels appear to increase early after breakdown in the alveolar epithelium. SP-A has been shown to be present in abnormal amounts in the BAL fluid of patients with IPF [22]. In a recent study, Kinder et al. [23] demonstrated that serum SP-A level at the time of diagnosis of IPF is a strong independent predictor of time to death or to lung transplantation, particularly during the first year of follow-up. Increased serum SP-A concentrations may identify a subset of patients with more “active” disease that increases the risk of death in the following year and was not predicted by other baseline, noninvasive clinical predictors, such as lung function test results.
Acute exacerbation of IPF: radiological/pathological aspects and pathogenesis
Acute exacerbation of IPF (IPF-AE) is a challenging problem, defined as a rapid deterioration of IPF during the course of the disease that cannot be attributed to infection, pulmonary embolism or heart failure; mortality rates may be as high as 60 to 70% over 3 to 6 months [24]. There is no known effective therapy for this devastating disease, and potential new approaches [25] in well-designed clinical trials are urgently needed. The pathological hallmark of IPF-AE is diffuse alveolar damage (DAD) superimposed on the UIP pattern characteristic in IPF [26]. IPF-AE can occur at any time during the disease course, and the risk of an exacerbation does not appear to be linked to the level of pulmonary function derangement, age or smoking history [27]. The histological findings from lung biopsy specimens show variable aspects; the typical UIP pattern is associated with signs of acute lesions, such as DAD with or without hyaline membranes, numerous fibroblastic foci, organising pneumonia, and haemorrhage with capillaritis [26, 28]. Churg et al. [29] described that three microscopic patterns of acute lung injury were seen in IPF-AE: DAD, organising pneumonia and a pattern of numerous very large fibroblastic foci superimposed on underlying fibrosis, and that patients with organising pneumonia or extensive fibroblastic foci as the acute pattern seem to do better than those with DAD. New areas of ground-glass opacities can be identified on HRCT during IPF-AE, and the extent and pattern of these are linked to prognosis [30]. In the IPF-AE, two basic computed tomography patterns may be classified: new areas of parenchymal opacification mainly within the peripheral region with relatively limited damage, and new areas of parenchymal opacification that spread rapidly throughout the lung with a fulminant clinical course. Akira et al. [30] demonstrated that survival was worse for patients with HRCT diffuse pattern than for those with both peripheral and multifocal pattern. Computed tomography scans of multifocal pattern were obtained in the earlier phase of acute exacerbation compared with those of diffuse pattern and multifocal pattern evolved into diffuse pattern for a short period. Multifocal pattern is considered to be the early phase of diffuse pattern. Peripheral pattern does not evolve into diffuse pattern for a short period. On the basis of computed tomography–pathological correlation in this study, diffuse pattern corresponded to DAD, whereas peripheral pattern mainly correlated with organising pneumonia or numerous fibroblastic foci. The computed tomography extent and patterns had a higher predictive value regarding patient survival than did the clinical and laboratory data [30].
Little is known about the pathogenesis of IPF-AE. Along with histopathology of DAD, there is evidence of loss of alveolar epithelial cell integrity [28]. It has been suggested that IPF-AE may represent a response to a clinically occult infection [31, 32] but direct evidence of an association with infections is still missing [33]. To generate new hypotheses regarding the molecular events that underlie IPF-AE and to identify new potential biomarkers for this syndrome, a recent study analysed the global gene expression patterns in the lungs of patients undergoing IPF-AE and compared them with stable IPF and control lungs [34]. Compared with control samples, IPF and IPF-AE lungs exhibited similar gene expression signatures. However, on direct comparison of IPF and IPF-AE the study identified differentially expressed genes and chose to focus the validation on CCNA2 and α-defensins. CCNA2, a general regulator of the cell cycle, was among the most upregulated genes in IPF-AE. Increased CCNA2 protein expression was localised to proliferating epithelial cells but not to mesenchymal cells. The overexpression and localisation of CCNA2 to epithelial cells but not to mesenchymal cells suggests that IPF-AE is probably an extension of the epithelial injury and dysregulation that characterises IPF [35] and definitely is not a result of uncontrolled fibroblast proliferation. Gene expression patterns indicate that IPF-AE represents an extension of the molecular process that underlies IPF and not a new process. Gene expression levels of α-defensins were upregulated in IPF-AE and their protein expression was localised to the alveolar epithelium in IPF-AE. Plasma α-defensin concentrations were higher in patients with IPF-AE compared with those with stable IPF or control subjects. Taken together, these results indicate the central role of the pulmonary epithelium in IPF-AE and suggest a potential role for α-defensins as peripheral blood biomarkers in IPF-AE [34].
New insights in therapy and pathogenesis of IPF
IPF is among the most therapeutically challenging lung disorders. No pharmacological agent improves survival in IPF and the search for an effective agent to treat IPF continues. Immunosuppressive treatment regimens have been used in the treatment of IPF; however, there is little evidence to support their efficacy and safety. As more has been discovered about the pathophysiological processes underlying IPF, new therapeutic approaches have been explored and controlled clinical trials conducted on these new therapies. For example, there is accumulating evidence that endothelin (ET)-1, a potent vasoconstrictor, plays an important role in the aetiology of IPF and that drugs that target ET-1 may be of benefit in patients with IPF. Bosentan, an oral dual receptor antagonist of ET-1, represents an interesting prospect for evaluation as a treatment for IPF. The randomised, placebo-controlled BUILD (Bosentan Use in Interstitial Lung Disease)-1 trial evaluated the efficacy, safety, and tolerability of bosentan in patients with IPF without evidence of pulmonary hypertension (PH) [36]. Although bosentan did not affect the exercise capacity of patients in this trial, as measured by a modified 6MWT, it demonstrated promising effects on time to disease progression or death, with a more pronounced treatment effect in the subset of patients entered on the basis of biopsy-confirmed IPF. These observations are being investigated further in the randomised, placebo-controlled, event-driven morbidity/mortality study, BUILD-3. Results of this study have been disclosed recently. While there was a consistent trend in favour of bosentan, the primary end-point, time to disease worsening or death, was not met. Studies with macitentan, a new, potent (∼10 times more potent than bosentan) dual receptor antagonist of ET-1, and ambrisentan, a more selective ET-1 receptor antagonist, are ongoing.
The effect of interferon (IFN)-γ in patients with IPF was also assessed in the multicentre, double-blind, randomised INSPIRE study with survival time as the primary end-point [37]. However, this study was stopped prematurely following a planned interim analysis. The results showed that overall survival had crossed the predefined stopping boundary for lack of benefit, although among the 826 randomised patients there was not a statistically significant difference between treatment groups in overall mortality (14.5% in the IFN-γ group compared with 12.7% in the placebo group).
Raghu et al. [38] report an important, albeit negative, result in a well-designed randomised controlled trial of tumour necrosis factor (TNF)-α antagonist therapy in patients with IPF. The negative result of the etanercept study is unequivocal because the trialists used a placebo-controlled design, which is ethically acceptable when no known effective therapy exists. It would be less appropriate to conduct a trial in which both groups receive therapy of unknown efficacy (even though standard of care); this study design makes results regarding the experimental intervention impossible to interpret. It remains unclear, for example, whether N-acetylcysteine has a beneficial effect in patients with IPF because both groups received prednisone and azathioprine [39]. The placebo-controlled design of the etanercept trial is an important benchmark for future IPF studies [40]. Because this study included only a modest number of subjects, it was not altogether surprising that there were no statistically significant differences between treatment groups in the three primary end-points: changes in FVC % predicted, DL,CO haemoglobin % predicted and alveolar–arterial oxygen tension difference (at rest) at 48 weeks. However, a consistent trend in favour of etanercept was observed in several lung function and quality-of-life measures. This study was limited by a lack of power, which minimised the chance of achieving a positive result. The results observed here support further investigation of TNF antagonists in clinical trials with adequate sample size and controls [38]. In a randomised, placebo-controlled trial of patients with mild to moderate IPF followed for 96 weeks, imatinib did not affect survival or lung function [41]. A randomised, controlled pilot trial compared oral cotrimoxazole to a placebo in 20 patients with advanced progressive fibrosing lung diseases over a period of 3 months, followed by 6 weeks of pulmonary rehabilitation before decoding [42]. Positive findings were reported for the shuttle-walking test as well as for lung function measures and dyspnoea score. The mechanism by which co-trimoxazole may improve or stabilise idiopathic interstitial pneumonia (IIP) is unknown but several possibilities exist: 1) antibacterial action: subclinical infection may influence inflammatory mechanisms and the fibrotic process; 2) antifungal actions: co-trimoxazone is active against fungi and protozoans, including pneumocystis, and many patients with IPF experienced long-term immunosuppression; and 3) immune modulating: co-trimoxazole is an effective monotherapy for the treatment of Wegener's granulomatosis and its anti-staphylococcal action and anti-inflammatory properties are hypothesised important mechanisms. Despite the many limitations of this preliminary pilot study, the utility of antibiotic or antifungal therapy as adjunctive therapy for advanced fibrotic lung disease may merit further investigation. Similar to other chronic pulmonary diseases (e.g. emphysema or even PH), there is emerging evidence that pulmonary rehabilitation may improve exercise tolerance, health status and quality of life in patients with diffuse parenchymal lung diseases (DPLDs) [43–45]. These studies were limited, however, by the small numbers of patients enrolled and the lack of any benefit beyond 6 months [44]. Adequately designed and powered studies are needed to confirm these initial results and to assess the contribution of rehabilitation as part of a comprehensive treatment approach to patients with DPLDs. Publication of results of multicentre, double-blind, placebo-controlled randomised study on pirfenidone treatment in IPF (CAPACITY trial) are pending. A total of 779 patients were enrolled in the CAPACITY trials at 110 sites in 11 countries. Preliminary results show a significantly smaller loss of lung function after 72 weeks than placebo in one study (CAPACITY 2), but the other almost identical trial (CAPACITY 1) did not show such benefit in the primary end-point; supportive evidence of a pirfenidone treatment effect was observed on a number of secondary end-points in CAPACITY 1 [46]. Data from these two studies suggests that pirfenidone has a positive treatment effect on patients with IPF. The CAPACITY trials follow a phase III clinical study conducted in Japan which demonstrated the ability of pirfenidone to reduce the decline of lung capacity and improve progression-free survival [47] and served as the basis for the Japanese regulatory authorities' approval of the drug for the treatment of IPF in Japan. The Food and Drug Administration evaluation of pirfenidone for IPF treatment in the USA is ongoing.
Despite a large number of new therapeutic agents being evaluated in clinical trials, there is no effective treatment for IPF patients. The recurrent and frustrating observation that steroids and immunosuppressants are of limited help in IPF, coincident with new insights into genetic aberrations in familial forms of IIP, caused a paradigm shift with respect to the pathogenetic mechanism of IPF: what was once believed to be triggered by chronic inflammation is now believed to be caused by repeated alveolar epithelial cell injury. Like many other complex human diseases, a single aetiology or aberrant signalling pathway may not be causal in all cases [48]. As its name suggests, IPF is a disorder with an enigmatic pathogenesis. It is currently believed that IPF is an epithelial–fibroblastic disease, in which unknown endogenous or environmental stimuli disrupt the homeostasis of alveolar epithelial cells, resulting in diffuse epithelial cell activation and aberrant epithelial repair [49]. It is already known that familial IPF occurs as an autosomal dominant disorder with variable penetrance [50–52]. To date, only three candidate genetic mutations have emerged from studies of pulmonary fibrosis in affected families. The evidence for genetic factors in sporadic IPF is much less compelling. Loyd [53] explores mutations in the genes encoding SP-C (SFTPC) and components of the telomerase gene, i.e. human telomerase reverse transcriptase and telomerase RNA component. Further research in this field will surely reveal other important candidates and will one day help to identify future therapeutic targets for this hugely challenging disease. Cronkhite et al. [54] describe the unexpected and fascinating finding that telomeres are severely shortened in peripheral leukocytes in approximately one-quarter of sporadic and familial cases of pulmonary fibrosis. In families with IPF with a genomic telomerase mutation as the basis, the parenchymal lung cells have a mutant telomerase (an enzyme that is responsible for maintenance of telomere lengths in replicating stem/progenitor cells), suggesting the possibility that cell demise or limited regenerative capacity, or both, may be a central feature that may initiate the disease process. Alder et al. [55] showed that telomere length in alveolar epithelium and peripheral blood leukocytes from patients with IPF was significantly shortened. Shortened telomeres in these patients occurred even in the absence of a family history or detectable mutations in telomerase. The striking loss of cellular homeostasis in IPF is notable for the increase in the number of mesenchymal cells, including clusters of myofibroblasts within the architecturally remodelled alveoli of the UIP lung. IPF is primarily a disease of the elderly, with a remarkable increase in incidence and prevalence beyond the fifth decade of life [56]. In the lung, the alveolar epithelial lining composed primarily of flattened, differentiated alveolar type 1 (AT1) cells comprise >95% of the alveolar surface area and are replenished by its tissue- and lineage-specific progenitors, the alveolar type 2 (AT2) cells. Lung-resident mesenchymal stem/progenitor cells (MSCs) have only recently been identified, both in adult lung of humans [57] and mice [58, 59]. As is true for adult tissue-resident stem/progenitor cells (but not differentiated somatic cells), both AT2 cells [60] and MSCs [59] express telomerase. Telomere shortening is associated with reduced capacity for stem cell renewal, cellular senescence, and organism aging [61]. One might predict that telomere shortening in AT2 cells would diminish regenerative capacity of the alveolar epithelium. Another intriguing question that the study raises is why the same host and environmental factors that presumably suppress the regenerative capacity of epithelial stem cells do not affect the mesenchymal response. This may be explained by the observation that telomere length and telomerase activity in stem/progenitor cells may be cell specific and autonomous [62]. In fact, it is likely that cells within even the same stem cell compartment may be influenced to varying degrees by the cumulative, stochastic events that influence telomere length and resultant regenerative capacity. This may also provide a plausible explanation for the observed spatial–temporal heterogeneity in UIP. Further studies are required to define telomerase expressing cells in the lungs of patients with IPF as well as telomere (dys)function in different stem cell compartments and their resultant cellular phenotypes. Such studies will aid in defining whether IPF represents a disorder of stem cell senescence and impaired lung regeneration [63].
In a recent study, it was demonstrated for the first time that a severe endoplasmic reticulum stress response in the AT2 cells seems to underlie the execution of the intrinsic apoptosis pathway, and thus the programmed cell death of this cell type, in patients with sporadic IPF. Endoplasmic reticulum stress response may represent an important trigger mechanism of the aberrant fibrotic repair observed in IPF [64].
IPF is characterised by myofibroblast accumulation and progressive lung scarring. Mesenchymal cells are responsible for deposition of extracellular matrix proteins such as collagen and fibronectin in the lung parenchyma and this compromises gas exchange. These mesenchymal cells may arise from three distinct sources. First, resident lung fibroblasts proliferate and differentiate into myofibroblasts, prodigious producers of collagen and contractile contributors to alveolar collapse and traction bronchiectasis. The second source may involve epithelial to mesenchymal transition, a process whereby epithelial cells release from basement membrane and undergo reprogramming that allows them to acquire a mesenchymal phenotype [65]. The third source involves recruitment of circulating bone marrow-derived precursors, known as fibrocytes, which share mesenchymal and leukocyte markers [66]. Significant increases in percentages of circulating fibrocytes were found in both stable and acute cohorts with IPF when compared with healthy volunteers, but the numbers were most striking in patients with IPF-AE [67]. This study suggests that the percentage of fibrocytes in circulation may serve as a biomarker for the presence of fibrotic lung disease and may be a useful biomarker for acute exacerbations [67]. Currently, therapeutic trials in IPF are hampered by the lack of robust easily obtained outcome variables for analysis and response to therapy. Changes in FVC are used to predict disease severity but may not reflect pathological changes in the disease process [3]. Serial analyses of fibrocytes may predict stability, deterioration and/or acute exacerbation. Additionally, this recent study indicates that fibrocyte percentages >5% are independent predictors of early mortality in IPF [67]. An ancillary study for the IPF Network, “prednisone, azathioprine and N-acetylcysteine: a three arm study that evaluates responses in IPF”, or PANTHER trial, will include an analysis of fibrocyte numbers and chemokine receptor profiling at baseline, mid-point, and at the conclusion of the study (weeks 0, 32, and 60) for up to 130 patients in each of three study arms [68].
Normal lung development and alveologenesis, which may continue into adolescence, are highly complex and regulated processes that are dependent on epithelial/endothelial–mesenchymal interactions. Although dormant in the uninjured adult lung, many developmental pathways are reactivated in the context of injury, repair and regeneration [69–71]. Together, these studies highlight the role of conserved developmental pathways in adult lung regeneration and support potential opportunities to modulate the reparative process in patients with DPLD.
NONSPECIFIC INTERSTITIAL PNEUMONIA
NSIP is often a source of diagnostic confusion. A recent study clearly demonstrates that NSIP is a separate entity and nicely determines its characteristics [72]. Key features of NSIP on HRCT are bilateral, symmetrical, predominantly lower lung reticular opacities with traction bronchiectasis and lower lobe volume loss that is usually diffuse or sub-pleural in the axial dimension but sometimes spares the sub-pleural lung [72]. If typical HRCT features are sufficient to allow a confident diagnosis of IPF/UIP in >50% of suspected cases, the HRCT sensitivity and specificity in the NSIP diagnosis are lower; there is overlap between the HRCT findings of UIP and those of NSIP in several cases. It must be pointed out that a confident diagnosis of NSIP requires surgical biopsy. A large proportion of patients initially considered to have idiopathic NSIP were subsequently reclassified as having hypersensitivity pneumonitis, IPF or organising pneumonia. When an HRCT scan shows classical features of hypersensitivity pneumonitis or organising pneumonia, these diagnoses are favoured over idiopathic NSIP, even if a surgical lung biopsy shows histological features of the NSIP pattern. Similarly, an HRCT showing a typical pattern of UIP (in particular, honeycomb changes) leads to a diagnosis of IPF, even when a surgical lung biopsy shows histological features of NSIP [72]. The difficulties inherent in trying to identify a discrete clinical, histological and radiological phenotype for NSIP arise because NSIP represents the common histological lesion of a range of diseases. NSIP is one of the most common patterns of interstitial pneumonia, not only in IIP, but also in patients with other disorders causing interstitial pneumonia, such as connective tissue diseases (CTDs), infection and drug or environmental toxicity. Kinder et al. [73] reported that 88% of their cases of idiopathic NSIP had manifestations of an undifferentiated CTD. In another study, ∼10% of the patients developed CTD during follow-up and they were younger and had higher rheumatoid factor positivity [74]. It was also found that honeycombing on the initial HRCT, initial pulmonary function test and changes in FVC at 12 months have considerable prognostic value [74]. Whether idiopathic NSIP is an autoimmune disease needs further investigations. In cases in which computed tomography or biopsy are believed to show features of NSIP, but in which there are incomplete data or suspicion of an alternative diagnosis, such as hypersensitivity pneumonitis, the term “NSIP pattern” is appropriate [72]. Therefore, it is current opinion that the integrative approach of clinical–radiological–pathological data must be extended to the whole spectrum of DPLD, as in all these disorders diagnosis is very complex, and the same problems of sampling error and interobserver variation between pathologists are encountered [72].
CONNECTIVE TISSUE DISEASE
A recent study confirmed that patients with CTD–interstitial pneumonia survived longer than those with IIP, but this was mainly because the survival of patients with CTD/UIP was longer than those with IPF/UIP, and not just due to a higher prevalence of a NSIP pattern in patients with CTDs (figs 4⇓ and 5⇓) [75]. In patients with a NSIP pattern as an overall group, there was no survival benefit in the CTD/NSIP group compared with the idiopathic NSIP group. Interestingly, mortality in UIP was higher in rheumatoid arthritis than in other CTDs and was only marginally lower than in IPF, although the small size of the rheumatoid arthritis subgroup is an important caveat [75]. UIP was more prevalent than NSIP in this rheumatoid arthritis cohort, and, based on earlier reports [76–78], appears to be much more prevalent in rheumatoid arthritis than in other CTDs.
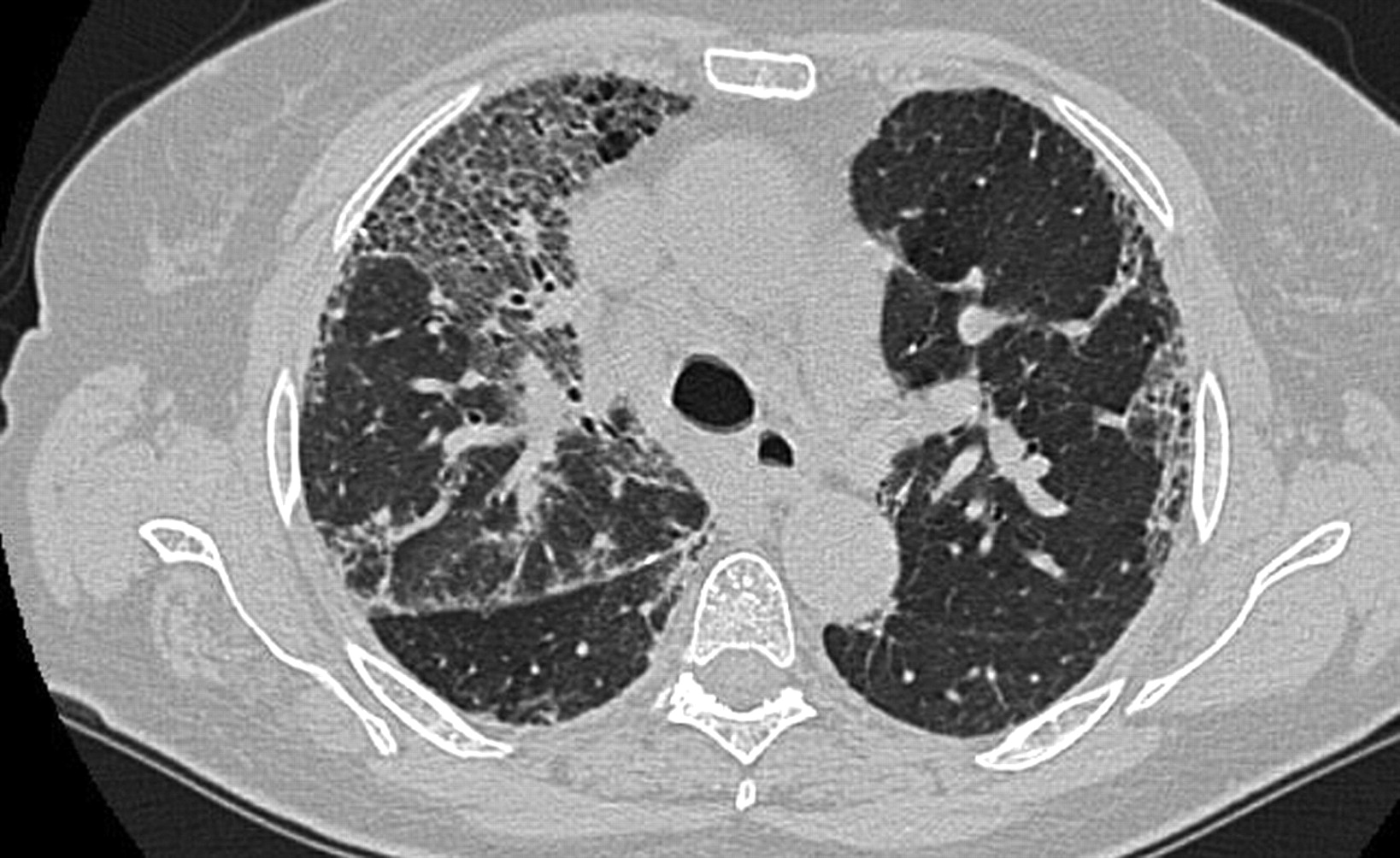
High-resolution computed tomography with nonspecific interstitial pneumonia pattern in a patient with connective tissue disease; bilateral reticulation and ground-glass are present. Although the abnormality shows peripheral predominance in about one-third of cases, the relative sparing of the immediate sub-pleural zone of lung seen in ∼20% of cases may sometimes be a helpful diagnostic finding.

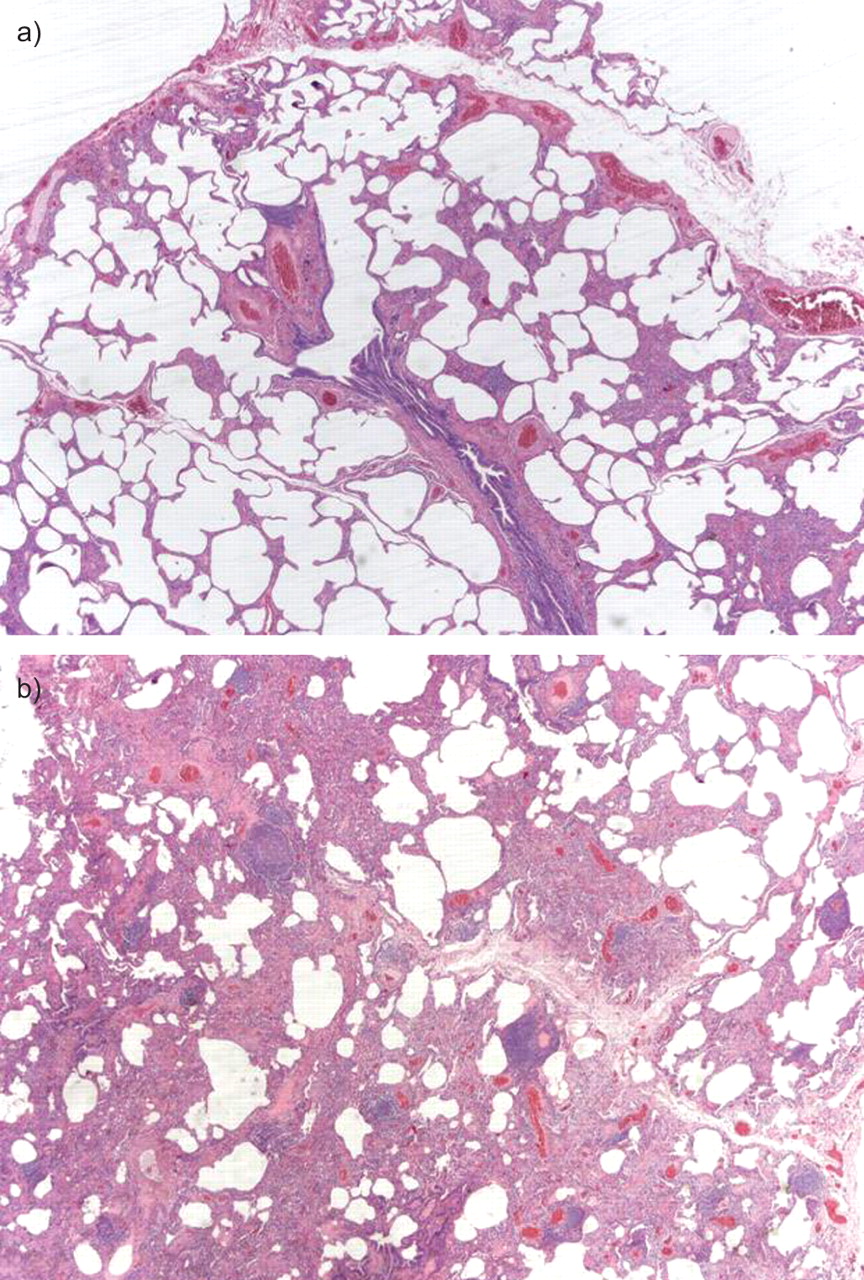
Nonspecific interstitial pneumonia fibrosing pattern in a case of connective tissue disease. a) The alveolar walls are thickened by interstitial fibrosis and chronic inflammation. b) Hyperplasia of the lymphoid tissue is present.
The Scleroderma Lung Study group reported that, after 1 yr of placebo-controlled oral cyclophosphamide for pulmonary fibrosis in systemic sclerosis (SSc), the benefits of active treatment persisted at 6 months but not at 1 yr [79]. Importantly, the treatment benefit resulted in the prevention of disease progression in more advanced fibrotic disease, rather than regression of reversible inflammatory disease. One obvious implication is that oral cyclophosphamide therapy should be consolidated by a less toxic agent in the longer term. Intravenous cyclophosphamide and oral mycophenolate mofetil have both been proposed as suitable longer term treatments, or even as substitutes for the initial use of oral cyclophosphamide [80–83]. Although historically considered to provide useful prognostic information in SSc, the presence of a BAL neutrophilia was recently found in a large SSc cohort to be linked only to early mortality and not to long-term survival or the rapidity of pulmonary function decline [84]. In a prospective study of 105 patients, only diffuse SSc was predictive of a decrease in pulmonary function; this observation does not support preliminary data suggestive of a causative role of oesophageal involvement [85]. In a recent study, serum levels of SP-D and KL-6, a mucin-like glycoprotein, appear to be indicative of “alveolitis” in SSc patients, and are significantly higher than in SSc patients without “alveolitis” [86]. Serum SP-D and KL-6 may serve as noninvasive serological means of assessing ILD in patients with SSc.
Hypersensitivity pneumonitis
Hypersensitivity pneumonitis, also termed extrinsic allergic alveolitis, is a form of DPLD resulting from an aberrant immunological response to an inhaled allergen in sensitised patients. Currently, no “gold standard” exists for diagnosing hypersensitivity pneumonitis, although identifying an inciting antigen linked to exacerbation and remission of symptoms is essential. A subset of patients with suspected hypersensitivity pneumonitis lacks an identifiable antigenic exposure. Lung biopsy plays an especially important role in identifying patients for whom no offending antigen is discovered, particularly those with chronic, persistent symptoms. Histological features that allow confident recognition of hypersensitivity pneumonitis in the appropriate clinical context include a bronchiolocentric interstitial pneumonia comprising a combination of chronic inflammation and variable degrees of fibrosis, chronic bronchiolitis and poorly formed granulomas that typically take the form of isolated multinucleated giant cells within peribronchiolar interstitium [87]. Occasionally, surgical lung biopsies may lack diagnostic features, showing instead NSIP without associated granulomatous inflammation. UIP also has been described in patients with suspected hypersensitivity pneumonitis [88]. Surgical lung biopsy is helpful in separating hypersensitivity pneumonitis from other forms of DPLD, including IIP. Late stage hypersensitivity pneumonitis and UIP may be indistinguishable in some patients [89]. The presence and the extent of fibrosis on HRCT predicts survival in hypersensitivity pneumonitis patients. Hypersensitivity pneumonitis may resemble NSIP or UIP by both radiological and histopathological assessment, and these appearances likely portend worse prognosis [88, 90, 91]. Recently, Silva et al. [92] demonstrated that the computed tomography features that best differentiated hypersensitivity pneumonitis from NSIP and UIP were lobular areas with decreased attenuation, absence of lower zone predominance, and presence of centrilobular nodules (fig. 6⇓).

a) High-resolution computed tomography shows innumerable ill-defined centrilobular ground-glass opacity nodules, characteristic of sub-acute hypersensitivity pneumonitis. Note that there are also foci of relative sparing associated with air trapping. b) Evidence of fibrosis with irregular reticular opacities, traction bronchiectasis and architectural distortion in chronic hypersensitivity pneumonitis. A large cyst is present in the lower right lobe.
A recent study proposes that the clinical course of fibrotic hypersensitivity pneumonitis, like other forms of fibrotic lung disease, can be associated with idiopathic acute exacerbations of disease leading to rapid respiratory deterioration and predicting a poor outcome. Further investigations into the similarities between these fibrotic lung diseases and the (possible) common pathway of acute exacerbations may yield additional insights into this recently recognised syndrome and, possibly, a better understanding of the mechanisms of fibrosis involved in these different disease processes [93].
LYMPHANGIOLEIOMYOMATOSIS
Lymphangioleiomyomatosis (LAM) is a rare lung disease affecting females, with onset typically during the childbearing years. LAM can occur in females with tuberous sclerosis complex (TSC), an autosomal dominant disease characterised by neurological disease (i.e. seizures, intellectual disability and autism) and benign tumours of the brain, skin, heart and kidneys. About 30% of females with TSC have evidence of cystic lung disease on computed tomography scans and are believed to have LAM (fig. 7⇓). While the rate of progression is variable, most females survive between 10 and 20 yrs after diagnosis. Four natural history studies found that loss of forced expiratory volume in 1 s (FEV1) averaged 75–156 mL per year [94–97]. LAM varies in clinical features and rate of progression; this, together with an absence of clear prognostic factors, results in patients being given conflicting information about prognosis. DL,CO, which is abnormal in more patients than FEV1, may be a more sensitive indicator of early disease. Patients with LAM may have partially reversible airflow obstruction. A positive response to bronchodilators is associated with an accelerated rate of decline in pulmonary function [98]. Lung transplantation remains the only viable option for end-stage disease. Several potential treatment regimens directed at hormonal manipulation have proven largely ineffective and the link between female sex and LAM remains unexplained [96]. Pathologically, LAM is characterised by two distinct components: LAM cells and cystic areas of alveolar destruction. Smooth muscle (SM)-like LAM cells represent a unique type of mesenchymal cell, expressing SM-specific markers, continuously proliferating and invading the lung parenchyma. Although the origin of LAM cells remains unknown, some progress has been made in our understanding of the genetics and signal transduction mechanisms regulating SM-like LAM cell proliferation and migration. In the past few years, our understanding of the SM-like cells in LAM has greatly improved. Abnormal growth, proliferation and migration of atypical SM-like LAM cells in the lungs lead to cystic destruction of the lungs. TSC1/TSC2 tumour suppressor complex mutations had been linked to the abnormal growth and migration of LAM cells. TSC2 dysfunction in LAM cells results in the constitutive activation of p70 S6 kinase (S6K1), which identifies S6K1 as a molecular target for treatment of LAM disease [99–102]. In summary, it is apparent that LAM cell behaviour, including growth, proliferation, adhesion and motility, plays a critical role in LAM disease. However, whether only TSC1 or TSC2 dysfunction accounts for the multisystem pathological changes seen in pulmonary LAM needs to be established. LAM pathology provides clues to the enigmatic complexity of LAM disease, the solving of which may lead not only to elucidating cellular and molecular mechanisms but may also pave the way toward finding the cure for LAM. The importance of further investigations into a role of SM-like cells in the aetiology and pathology of LAM cannot be overestimated, not only because they may uncover mechanisms of LAM pathology but may also have the potential to develop novel therapeutic strategies for LAM treatment [102]. The role of mammalian target of rapamycin (mTOR) inhibition in the therapy of patients with TSC and LAM was recently investigated [103]. These rapid-fire advances led to a prospective therapeutic trial in patients with LAM and TSC with angiomyolipomas, with angiomyolipoma volume as the primary end-point. Patients received 12 months of rapamycin, followed by 12 months of follow-up after discontinuation of rapamycin [104]. There was no randomisation or placebo control arm in this initial study. Of 25 males and females enrolled, 20 patients completed 12 months of rapamycin and 18 patients completed the full study, including the 12 months of follow-up after rapamycin. Angiomyolipoma volume decreased to 53% of baseline after 12 months of rapamycin therapy, and after discontinuation increased to 86% of baseline at 24 months. 18 of the patients had LAM, with 12 having TSC-associated LAM and six having sporadic LAM. The improved lung function in the 10 females is extremely encouraging, and perhaps unexpected: one might have expected stabilisation of lung function, rather than improvement, because the degradation of lung parenchyma that accompanies LAM would not likely be correctable, at least during 12 months of therapy with a TORC1 (mTOR complex 1) inhibitor. Although the mechanisms of this improvement are unknown, one possibility is that shrinkage or elimination of LAM cells improves the elasticity and mechanics of the residual areas of normal lung. Importantly, however, a second study in the UK has reported interim results of a 24-month trial of rapamycin for patients with TSC and LAM [105], and found no evidence of improved FEV1 or FVC in the four females with LAM who had completed 12 months of rapamycin. Possible explanations for these differences include an unrecognised difference in the severity or clinical parameters of LAM between the Cincinnati and UK cohorts, which may be amplified by the small numbers of patients, the impact of dose reductions or cessations related to adverse events, and/or the possibility of an effort-dependent placebo effect on the results of pulmonary function testing. It is likely that clarity will be provided when the completed data are available from the UK trial and when results are available from the Multicenter International LAM Efficacy of Sirolimus (MILES) trial, the first prospective, randomised clinical trial in pulmonary LAM, representing another landmark in the history of LAM research [103]. This trial completed accrual in 2009. An open-label, nonrandomised, within-subject dose escalation safety, tolerability and efficacy study of everolimus (a second generation mTOR inhibitor) in females with sporadic or TSC-associated LAM is ongoing. A case report showed benefit from doxycycline in a patient with LAM [106]. This case supports the hypothesis that doxycycline may represent a promising therapy for LAM. The evaluation of the role of doxycycline in LAM therapy is ongoing. Recently, the European Respiratory Society published consensus guidelines elaborated from a core panel of specialists for the diagnosis and management of LAM [107]. The recently established International LAM Registry is a component of a set of web-based resources, including a patient self-report data portal, aimed at accelerating research in rare diseases in a rigorous fashion and demonstrates that a collaboration between clinicians, researchers, advocacy groups and patients can create an essential community resource infrastructure to accelerate rare disease research [108].

High-resolution computed tomography (HRCT) showing typical changes in a patient with advanced definite lymphangioleiomyomatosis: extensive cystic changes are present. HRCT shows numerous discrete, round, thin-walled lung cysts.
PH IN DPLD
PH is prevalent and clinically relevant in various DPLDs [109–111]. The incidence of PH in patients with IPF increases from 39% at the time of listing for lung transplantation to 78% at the time of transplantation [112]. Among the 70 patients who underwent right heart catheterisation (RHC) during their initial workup, the prevalence of PH based on standard criterion (mean pulmonary arterial pressure >25 mmHg) was 8.1% (six patients) [113]. Lettieri et al. [114] reported a markedly higher prevalence of 31.6% among IPF patients who underwent the evaluation while waiting for lung transplantation. In estimations of pulmonary arterial pressure by Nadrous et al. [115] using the ultrasound cardiography (USCG) method, systolic pulmonary arterial pressures >50 mmHg were present in 30.7% of patients. Differences in the populations and measuring methods in these three studies make it difficult to directly compare the prevalence of PH. Generally, the degree of PH in IPF is mild to moderate, with few subjects developing severe PH by the time of listing for lung transplant. Elevated concentrations of N-terminal pro-brain natriuretic peptide levels and low DL,CO [116] may be predictive of the presence of PH in DPLDs. Studies from the past few years [114, 115] have shown that PH measured with RHC or USCG in IPF patients is associated with low DL,CO, shorter walk distances and desaturation during exercise. Similar relationships have been identified when brain natriuretic peptide was used as a surrogate marker for PH in IPF patients [117]. The optimal approach to identifying which IPF patients have PH remains controversial, however. Nevertheless, increasing PH is associated with an increasing risk of death in IPF patients [114, 115], while elevated pre-operative PH may contribute to post-transplant mortality in IPF [118, 119]. Factors other than the extent of fibrosis appear to play a significant role in the genesis of PH in patients with IPF. These might include, but are not necessarily limited to, local or systemic hypoxaemia, pulmonary thromboembolism, cardiac dysfunction, genetic predisposition, and variable cytokine or chemokine expression. The aetiology and course of PH in IPF warrants further study, as do therapies directed at preventing or treating this common complicating comorbidity [120]. With the advent of newer options for treating PH, coupled with the lack of effective therapies for IPF, therapy for disproportionate PH (PH associated with DPLD with a median pulmonary arterial pressure level above the usual values) appears an attractive area of investigations.
One important point in recent years in the field of smoking-induced chronic lung disease has been the identification of the syndrome of combined pulmonary fibrosis and emphysema (CPFE) [121]. Not just a distinct phenotype of IPF, the syndrome of CPFE is characterised by the association of distinct features, including tobacco smoking, severe dyspnoea, unexpected subnormal spirometry, severely impaired DL,CO, hypoxaemia on exercise, and characteristic imaging features (centrilobular and/or paraseptal emphysema, and diffuse interstitial opacities suggestive of pulmonary fibrosis of the lower lobes). The pathological features have not yet been formally studied. Pathophysiology of the syndrome beyond the obvious role of tobacco smoking remains to be explored [122]. Suspected in patients with dyspnoea and basal crackles unexplained by spirometry, the syndrome of CPFE can be recognised by the presence of both “significant” emphysema and fibrosis features on HRCT of the chest (fig. 8⇓). Importantly, patients with the syndrome of CPFE have a high probability of severe pre-capillary PH [121], which carries a poor prognosis, with only 60% survival at 1 yr from the date of RHC [123]. Indeed, the risk of developing PH is notably higher in CPFE than in IPF without emphysema [124] and PH (and not only the presence of associated emphysema) represents the main independent determinant of mortality in patients with CPFE [121, 124].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
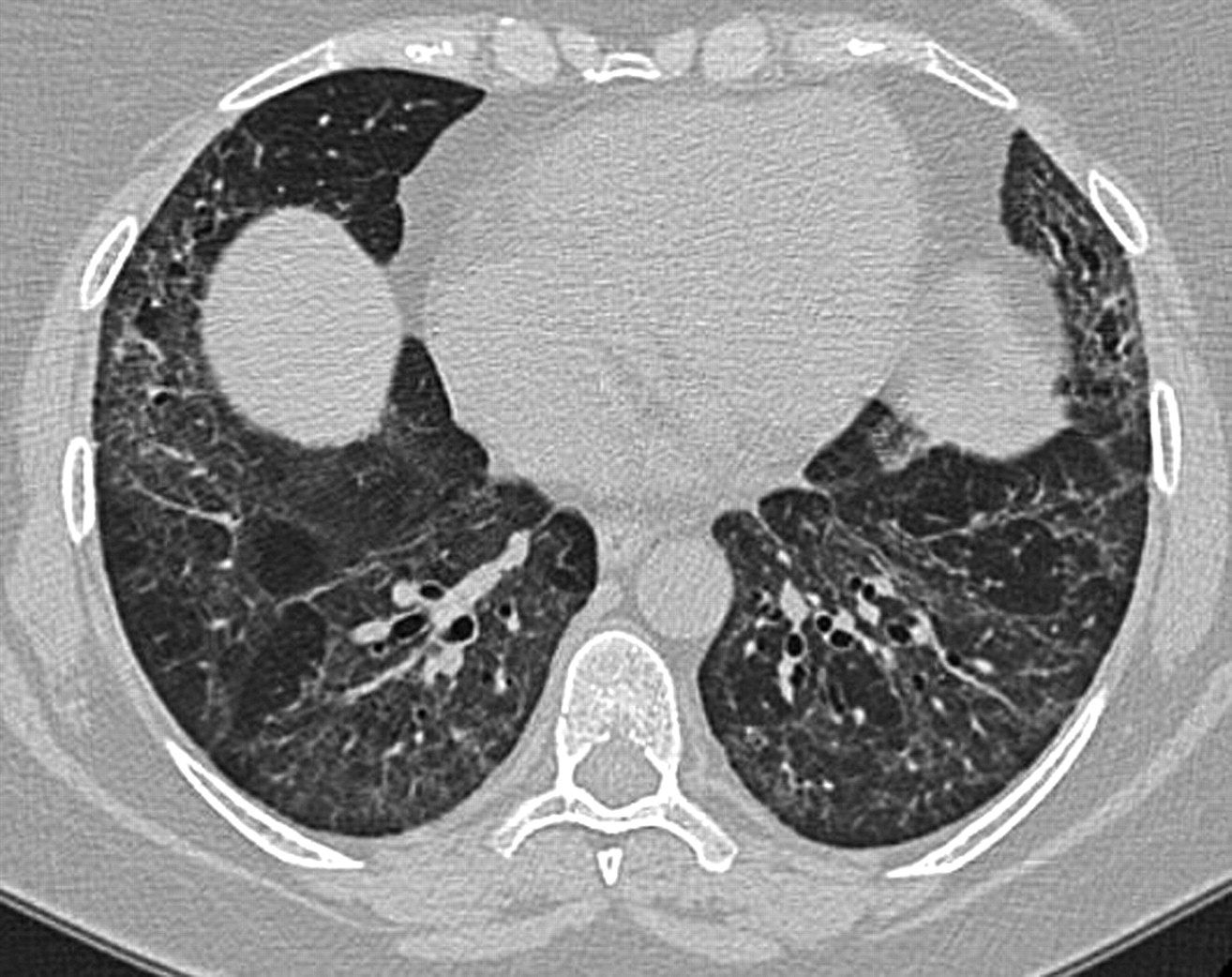
a) Significant emphysema in upper lobes and b) fibrosis in lower lobes in a patient with combined pulmonary fibrosis and emphysema.
Statement of interest
S. Harari received a grant of €2,000 from Actelion for participating on an advisory board in 2010.
Provenance
Submitted article, peer reviewed.
- Received March 13, 2010.
- Accepted March 29, 2010.
- © ERS
References









