





Abstract
Asthma is a complex, persistent, inflammatory disease characterised by airway hyperresponsiveness in association with airway inflammation. Studies suggest that regular use of high-dose inhaled corticosteroids and long-acting bronchodilators or omalizumab (a humanised monoclonal antibody that binds to immunoglobulin E and is often used as next-step therapy) may not be sufficient to provide asthma control in all patients, highlighting an important unmet need. Interleukin-4, interleukin-13, and the signal transducer and activator of transcription factor-6 are key components in the development of airway inflammation, mucus production, and airway hyperresponsiveness in asthma. Biological compounds targeting these molecules may provide a new therapeutic modality for patients with uncontrolled severe asthma. The purpose of this review is to summarise current studies of compounds targeting the interleukin-4/interleukin-13 pathway and to provide a rationale for the development of such compounds for this use.
- Asthma
- interleukin-4
- interleukin-13
- monoclonal antibody
- signal transducer and activator of transcription factor-6
Asthma is characterised by airway hyperresponsiveness (AHR) and inflammation, as well as underlying structural changes to the airways. Current treatment regimens are effective in controlling asthma in the majority of patients. Yet, nearly 30% of patients in the Gaining Optimal Asthma controL (GOAL) study failed to maintain asthma control despite regular use of high-dose fluticasone and salmeterol [1]. Omalizumab (Xolair®; Genentech Inc., South San Francisco, CA, USA), a humanised monoclonal antibody that binds to immunoglobulin (Ig)E, has been shown to decrease asthma exacerbations and corticosteroid requirements, as well as reduce hospitalisations and emergency room visits [2]. However, ∼40% of allergic asthmatics in the Investigation of Omalizumab in Severe Asthma Treatment (INNOVATE) study failed to achieve optimal clinical response to omalizumab [2]. Similarly, in patients treated with long-acting β-agonist (LABA)/inhaled corticosteroid (ICS) combinations, the addition of omalizumab versus placebo provided improvements in efficacy but resulted in a substantial proportion of incomplete responses [3]. These studies suggest that combination therapy with corticosteroids, LABAs and compounds blocking the IgE pathway may not be sufficient to provide optimal control of asthma in some patients, highlighting an unmet clinical need for atopic and nonatopic asthmatics.
Major determinants for the heterogeneity of severe asthma are atopy, lung function, age of asthma onset and duration, sex, symptoms, medication use and healthcare utilisation [4]. Interleukin (IL)-4 and IL-13, expressed by T-helper type 2 (Th2) cells, are key cytokines in the pathogenesis of atopy and atopic asthma [5, 6]. Both IL-4 and IL-13 promote acute inflammatory processes and underlying structural changes to the airways, and their receptors are expressed on a number of cell types [7]. Expression of IL-4 in the lung of IL-4 transgenic mice elicits an inflammatory response characterised by epithelial cell hypertrophy and accumulation of macrophages, lymphocytes, eosinophils and neutrophils, lacking an AHR response to inhaled methacholine [8]. Yet, Perkins et al. [9] have demonstrated that IL-4 can induce IL-13-independent AHR and goblet-cell hyperplasia in a mouse model of asthma. IL-13 induces AHR in addition to eosinophilic inflammation and mucus hypersecretion [10–14].
Elevated IL-13 levels have been detected in the airways and sputum of patients with asthma [15, 16] and mast cell-derived IL-13 and IL-4 have been observed in the airway smooth muscle cells of these patients [7, 17]. In addition to increased IL-13 levels, IL-4/IL-13 receptor (R) subunits are also abnormally expressed in this subgroup of patients [5].
Both IL-4 and IL-13, which share the IL-4Rα subunit in their cognate receptors, activate signal transducer and activator of transcription factor-6 (STAT-6) [7]. STAT-6 is required for IL-4 production from antigen-stimulated murine splenocytes [18]. Moreover, activation of STAT-6 is critical for the differentiation of naïve T-cells into Th2 effector cells, and STAT-6 regulates IL-4- and IL-13-induced production of Th2 chemokines, including eotaxin, from airway epithelial cells, fibroblasts and smooth muscle cells [19–23]. Thus, the IL-4/IL-13/STAT-6 pathway plays a key role in asthma pathogenesis.
Different approaches have been used to target the IL-4/IL-13/STAT-6 pathway, including soluble IL-4R compounds, neutralising antibodies specific for IL-4 or IL-13, antibodies or chimeric proteins targeting IL-4/IL-13R, and therapeutic compounds targeting STAT-6 and other downstream signalling molecules of IL-4/IL-13R.
THE IL-4/IL-13/STAT-6 SIGNALLING PATHWAY
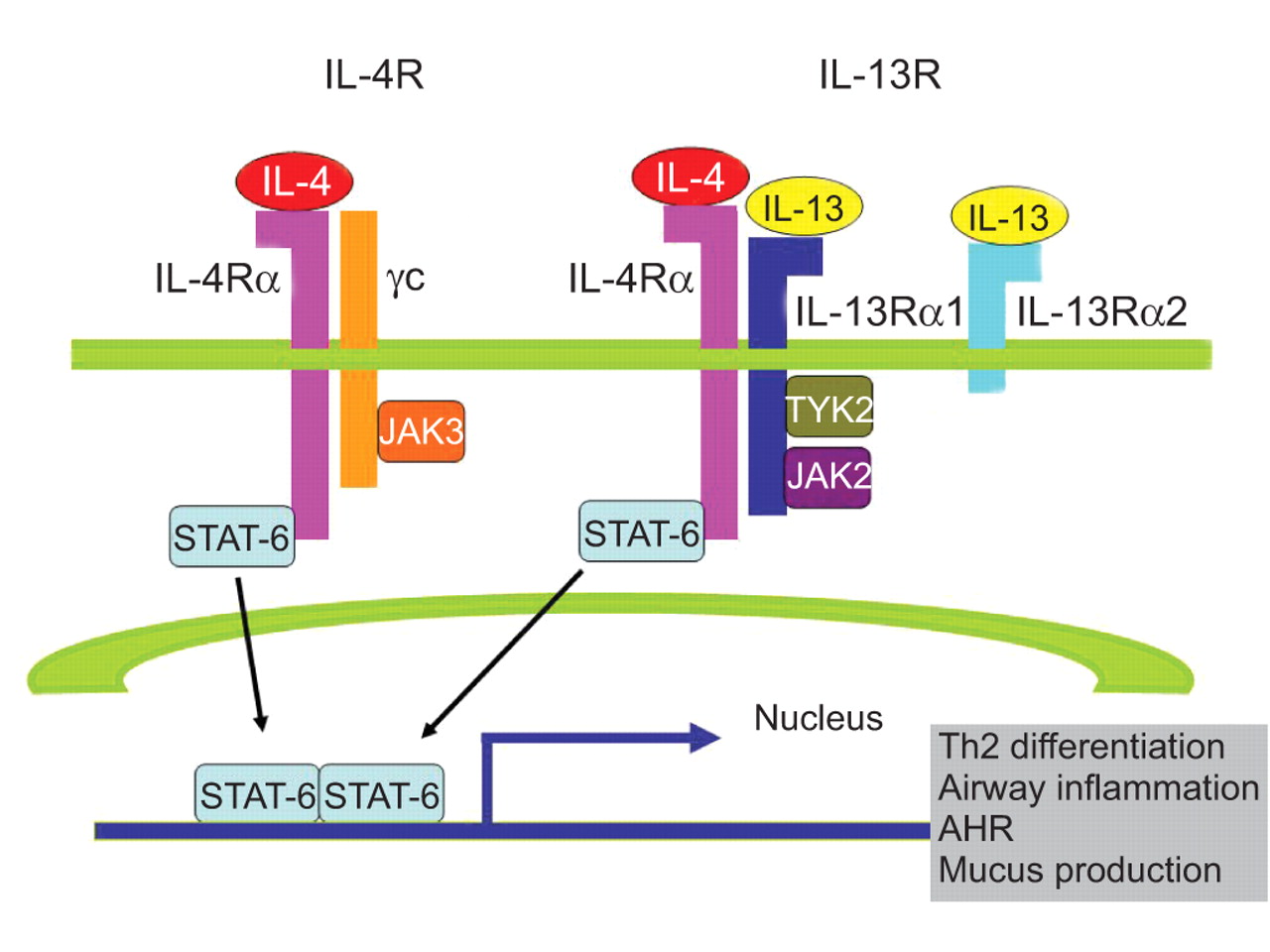
IL-13 functions are mediated by a complex receptor system, including type I (IL-4Rα/common γ chain (γc)/IL-4) and type II (IL-4Rα/IL-13Rα1/IL-4, IL-4Rα/IL-13Rα1/IL-13) receptor systems (table 1⇓) [24, 25]. IL-4 binds to the type I receptor, whereas both IL-13 and IL-4 bind to the type II receptor (fig. 1⇓), and differences exist in their signalling potency and kinetics [26]. Differences in signalling are due in part to the specific binding modalities of the two cytokines. IL-4 binds to the IL-4Rα subunit of the type II receptor complex, whereas IL-13 binds to the IL-13Rα1 subunit of the same receptor complex. Additional differences in biological effects result from the ability of the IL-4Rα subunit to dimerise with the γc subunit of the type I receptor complex, to which IL-13 is unable to bind [11].
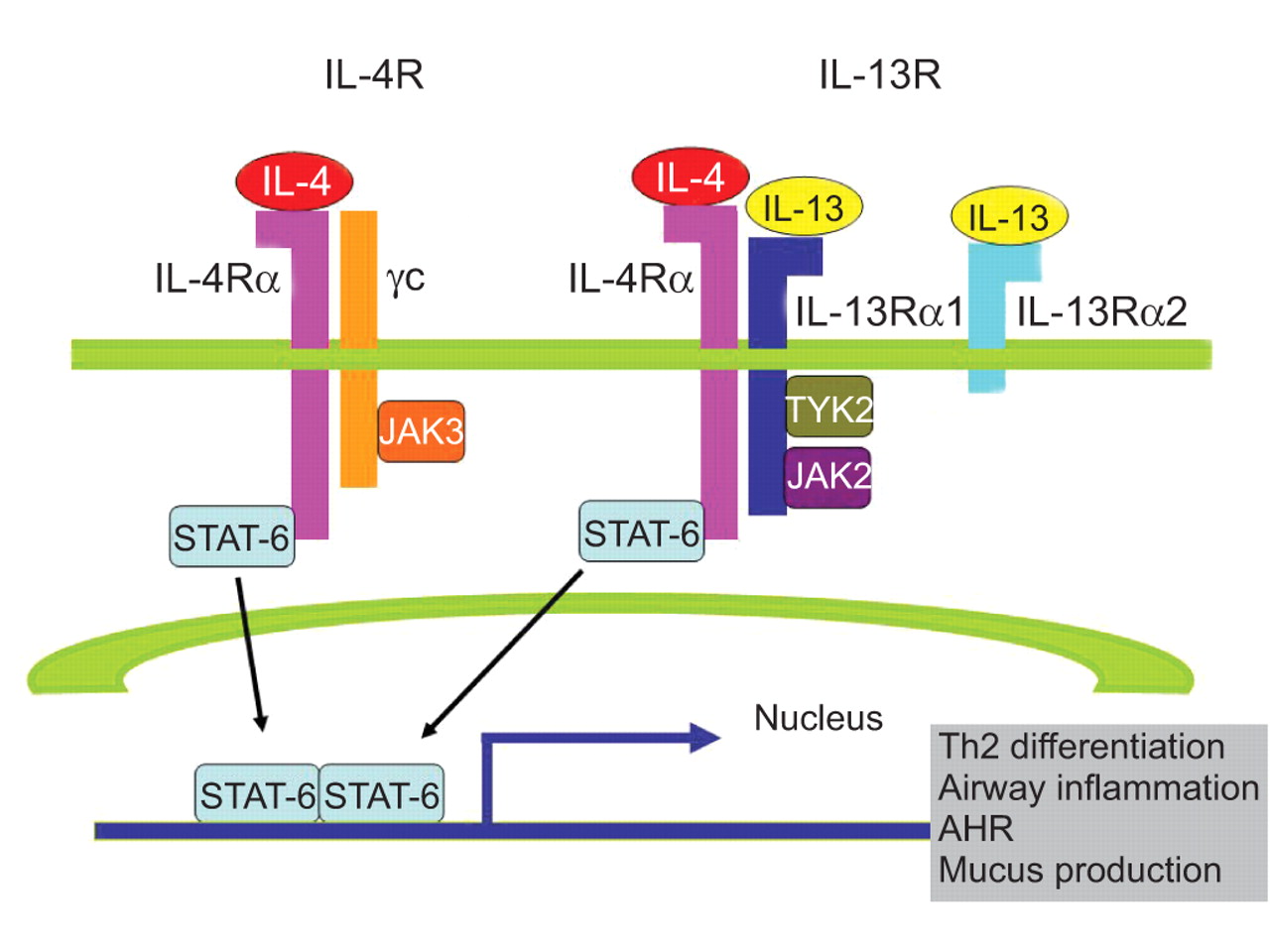
Schematic diagram of the interleukin (IL)-4/IL-13/signal transducer and activator of transcription factor (STAT)-6 signalling pathways [25]. Both IL-4 and IL-13 signal via the IL-4Rα, a component of the type I (IL-4Rα and γc) and type II receptors (IL-4Rα and IL-13Rα1). IL-4 signals via both type I and II receptor pathways, whereas IL-13 signals only via the type II IL-4R. IL-13 also binds to the IL-13Rα2 chain, which does not contain a transmembrane-signalling domain and is thought to act as a decoy receptor. γc activates janus kinase (JAK)3, whereas IL-13Rα1 activates tyrosine kinase 2 (TYK2) and JAK2. Activated JAKs then phosphorylate STAT-6. Phosphorylated STAT-6 dimerises, migrates to the nucleus, and binds to the promoters of the IL-4 and IL-13 responsive genes, such as those associated with T-helper type 2 (Th2) cell differentiation, airway inflammation, airway hyperresponsiveness (AHR) and mucus production.
The interleukin(IL)-4 and IL-13 receptors and their ligands
The γc subunit activates janus kinase (JAK)3, whereas IL-13Rα1 activates two other tyrosine kinases, tyrosine kinase 2 (TYK2) and JAK2 [11, 27]. Activated JAKs mediate the phosphorylation of the cytoplasmic tail of IL-4R on conserved tyrosine residues that serve as docking sites for proteins containing Src homology 2 (SH2) domains. Three closely clustered tyrosine residues serve as docking sites for STAT-6. The binding of IL-13 to IL-13Rα1 also activates STAT-6 because the IL-13:IL-13Rα1 complex binds with high affinity to IL-4Rα [28]. Consistent with the ability of each of these cytokines to activate STAT-6 signalling, the majority of the physiological manifestations in allergic disorders, including Th2 cell differentiation, AHR, mucus cell metaplasia, and IgE synthesis, are STAT-6 dependent. In contrast to the type II receptors that transduce intracellular signals, the IL-13R subunit, IL-13Rα2, is believed to be a decoy receptor because of its short cytoplasmic tail. IL-13Rα2 is present on the cell membrane, intracellularly, and in a soluble form. Cell membrane IL-13Rα2 receptors may have some signalling capabilities, but soluble IL-13Rα2 receptors are critical endogenous modulators of IL-13 responses and may, therefore, have a more complicated function than previously thought [29]. IL-13Rα2 is upregulated in cells with heightened responses to IL-13, and increased expression levels of the receptor subunits IL-4Rα and IL-13Rα2 have been reported in fibroblasts from surgical lung biopsies of patients with idiopathic interstitial pneumonia [30, 31].
IL-4/IL-13/STAT-6 PATHWAY GENETICS
In addition to upregulated levels of IL-13 and its receptor subunits, several single nucleotide polymorphisms (SNPs) of genes encoding for IL-13, its associated signalling molecules (STAT-6 and IL-13Rα1) and IL-4 have been linked to asthma and the atopic syndrome [32]. These polymorphisms cause aberrant binding and signalling activities that result in overactivation of the IL-4/IL-13 pathway. Among the SNPs associated with bronchial asthma or atopy, functional variants of IL-13 have been identified [33]. These variants have a lower affinity for IL-13Rα2 and enhanced stability, compared with their wild-type receptor counterparts, causing upregulation of IL-13 levels in vivo [34, 35]. Another IL-13 variant discovered has a minor allele frequency of ∼20% in the Caucasian population. This is the only nonsynonymous coding SNP found, to date, in IL-13, leading to a nonconservative substitution of a positively charged Arg residue at 110 in the mature polypeptide to a neutral Glu (R110Q).
In populations throughout the world, R110Q has been associated with high serum IgE titres, allergy and atopic dermatitis [36, 37]. The IL-13 residue 110 lies in the region of the molecule thought to interact with IL-13Rα1 and IL-13Rα2 [38]. Several hypotheses have been proposed to explain the association of this IL-13 polymorphic variant with the tendency to develop atopic disease, including decreased affinity for binding to the negative regulatory element (IL-13Rα2) [33], increased functional activity mediated through IL-13Rα1 [39] and enhanced stability in plasma [35]. In addition to the R110Q variant, associations have also been found between atopic susceptibility and the IL-13 promoter polymorphisms, -1055 C/T and -1112 C/T [36, 40]. Polymorphisms in genes encoding for IL-4Rα, IL-13Rα1 or STAT-6 [37, 40] may also predispose individuals to atopy development. For example, the substitution of Ile for Val (Ile50Val) and Arg for Gln (Arg576Gln) in the extracellular and cytoplasmic domain of the IL-4Rα gene, respectively, enhances signal transduction via IL-4R by augmenting the activation of STAT-6 and by impairing the binding of the negative regulatory molecule, protein tyrosine phosphatase SHP-1 [41, 42].
Collectively, these human genetic data strongly support a role for the IL-4/IL-13 pathway in contributing to the risk of developing atopic disease.
PRE-CLINICAL EVIDENCE FOR THE ROLE OF IL-4/IL-13/STAT-6 IN ASTHMA
Evidence supporting a link between the IL-4/IL-13/STAT-6 pathway and airway remodelling or fibrotic disease has been generated from studies in rodents treated with recombinant IL-13, compounds targeting IL-13 and its receptors, and compounds targeting genetic elimination of IL-13 [10, 12, 38–40, 43].
Inhibition of IL-13 through the use of antibodies [44, 45] or genetic deletion [46] has been shown to reduce AHR, eosinophil recruitment, mucus overproduction and development of airway remodelling by reducing fibrosis or excessive collagen deposition (fig. 2⇓), processes in which other proteins, such as matrix metalloproteinase (MMP)-9 or MMP-12, may also play an important role interdependently or independently of the IL-4/IL-13/STAT-6 pathway [47]. Pulmonary overexpression of IL-13 in rodents mimics many of the features of human asthma, such as lung eosinophilia, epithelial hypertrophy, mucus hypersecretion, AHR and airway remodelling [13]. In contrast to IL-13, IL-4 transgenic models do not exhibit significant airway fibrosis or AHR, supporting the hypothesis that IL-13 is a key mediator of many of the pathological features associated with the inflammation and airway remodelling characterising lung disease [8].

{kind=link}
{kind=link}
Schematic diagram of the potential cellular effects of interleukin (IL)-4/IL-13 on inflammatory and structural cells in asthma. Both IL-4 and IL-13, produced by T-helper type 2 cells and mast cells, promote airway inflammation by activating eosinophils, macrophages, and dendritic cells; airway remodelling by enhancing proliferation/activation of fibroblasts; immunoglobulin (Ig)E production by activating B-cells; mucus production by stimulating airway epithelial cells/goblet cells; and airway hyperresponsiveness by activating airway smooth muscle cells.
Anrukinzumab (IMA-638), a humanised monoclonal antibody specific for IL-13, has been shown to inhibit antigen-induced pulmonary inflammation in nonhuman primates. Bree et al. [48] have demonstrated that IL-13 blockade reduces lung inflammation following Ascaris suum challenge in cynomolgus monkeys [48]. Bronchoalveolar lavage (BAL) cells and BAL fluid were collected before and after the antigen challenge and assayed for cellular content by means of differential count. Eosinophil, neutrophil and total BAL cell counts were reduced in animals treated with anrukinzumab or a mouse antibody specific for human IL-13 (mAb13.2), compared with values measured in control animals who were untreated, administered saline or treated with a human IgG of irrelevant specificity. Additionally, levels of eotaxin and RANTES (regulated upon activation, normal T-cell expressed and secreted) in BAL fluid were reduced in anti-IL-13-treated animals, compared with those observed in controls.
Swart et al. [49] reported the effects of IL-4Rα blockade on lung inflammation and AHR in a murine model of cockroach allergen-induced asthma using the murine surrogate for AMG-317 (fully human IgG2 monoclonal antibody), Mu317RAXMu. Mediators of inflammation and inflammatory cell counts were decreased in BAL fluid, while inflammatory cell counts were also decreased in lung tissue. Serum IgE levels were elevated following the allergen challenge but returned to near baseline values following treatment. Also, AHR, as measured by methacholine challenge, was reduced.
Blanchard et al. [43] demonstrated the clinical activity of CAT-354, an anti-IL-13 monoclonal antibody, in a murine model of airway inflammation and AHR. BALB/c mice were treated with CAT-354, and human IL-13 was injected intratracheally. Human IL-13 induced dose-dependent AHR, airway eosinophilia and goblet cell metaplasia in control animals. However, mice pre-treated with CAT-354 showed significantly reduced AHR, airway eosinophilia and oesophageal eosinophilia.
A mouse soluble IL-4 mutant has been shown to have antagonist effects against both IL-4 and IL-13 in vitro via the formation of an unproductive complex with IL-4Rα [50]. Treatment of mice with this antagonist completely inhibited the humoral immune response to allergen and subsequent development of disordered lung function upon allergen challenge [50]. Similarly, BAY 16-9996 (AerovantTM; Aerovance, Berkeley, CA, USA) administered subcutaneously as a human IL-4 double mutein was effective in reducing airway inflammation and AHR in a primate model of asthma [51].
AIR645, a dual inhibitor of IL-4 and IL-13, is a 2'-O-methoxyethyl second-generation antisense drug targeting the mRNA encoding the IL-4Rα subunit. Mouse-optimised ISIS-369645 reduces IL-4R protein and mRNA levels. In murine models of asthma, ISIS-369645 reduced lung cytokine production, inflammation, and AHR [52]. Following inhalation, rapid distribution to the airways and therapeutic drug concentration levels were reached in multiple cell types, with little systemic exposure.
STAT-6 is required for the development of airway inflammation, mucus production and AHR in animal models of acute allergic airways disease [21, 53, 54]. McCusker et al. [55] generated a cell-penetrating STAT-6 inhibitory peptide (STAT-6-IP) consisting of the protein transduction domain 4 fused to a phosphotyrosine-containing sequence predicted to bind with high affinity to the SH2 domain of STAT-6; thus, acting as a dominant negative inhibitor of the wild-type STAT-6 protein. STAT-6-IP inhibits STAT-6-dependent production of IL-4 and IL-13 from antigen-stimulated primary murine splenocytes, as well as IL-4-induced eotaxin-3 production from cultured human bronchial epithelial cells (BEAS-2B). STAT-6-IP inhibited ovalbumin-induced production of IL-4 and IL-13 in vitro but did not affect production of interferon (IFN)-γ, demonstrating specificity for inhibiting Th2 cytokine production [55]. Furthermore, intranasal delivery of STAT-6-IP to murine models of allergic rhinitis and asthma inhibited ovalbumin-induced lung inflammation, mucus production, accumulation of eosinophils and IL-13 in BAL fluid, and ovalbumin-dependent AHR [55]. These data demonstrate that local application of cell-penetrating peptide inhibitors of STAT-6 has significant potential in the treatment of allergic rhinitis and asthma.
CLINICAL STUDIES
Targeting IL-4
Soluble IL-4R compounds used in clinical trials of asthma contain the extracellular portion of IL-4Rα, which binds IL-4 and thus inhibits its function [56]. Initial studies with soluble IL-4R were performed in subjects with mild-to-moderate persistent asthma who were withdrawn from their ICS therapy and then randomly assigned to receive placebo or single-dose nebulised IL-4R [56, 57]. Treatment with nebulised IL-4R improved asthma symptom scores, decreased β2-agonist metered-dose inhaler use, improved forced expiratory volume in 1 s (FEV1) and decreased methacholine airway responsiveness [56]. No significant adverse events or development of anti-IL-4R antibodies were detected. A subsequent phase I/II study in subjects with moderate, persistent asthma compared the effect of weekly nebulised IL-4R doses administered for 3 months to single-dose treatment of IL-4R [57]. The safety profile of nebulised IL-4R over 3 months was similar to that of placebo; however, a greater decline in FEV1 was observed in the placebo group (-13% predicted) compared with those receiving IL-4R treatment (-2% pred; p = 0.05) [57]. These initial studies suggest a potential advantage of IL-4R therapy in asthma, underscoring the importance of IL-4 in its pathogenesis. Yet, questions still remain about the efficacy of this therapy for the treatment of asthma [58].
Targeting IL-13
Evidence from animal models supported the use of a number of pharmaceutical IL-13 neutralising antibodies (table 2⇓) [10, 44, 45], including anrukinzumab, IMA-026, TNX650, QAX576, MILR1444, GSK-679586 and CAT-354 in phase I/II clinical trials of asthma.
Interleukin(IL)-4/IL-13 modulators
A phase II, randomised, double-blind, placebo-controlled study of anrukinzumab was completed in Canada in patients (n = 27) with mild, atopic asthma [61]. The primary study endpoint was maximum change in FEV1 post-allergen challenge during early and late asthmatic response (EAR and LAR, respectively), compared with baseline values. EAR and LAR area under the curve (AUC) values were significantly inhibited by anrukinzumab compared with placebo (EAR AUC 0–3 h, 46.3% inhibition versus placebo, p = 0.030; LAR AUC 3–7 h, 49.0% inhibition versus placebo, p = 0.039). No serious adverse events or changes in blood haematology, chemistry or vital signs were observed after treatment with anrukinzumab. These data suggest that IL-13 plays a significant role in early and late allergen-induced bronchoconstriction in humans. Another phase II, randomised, double-blind, placebo-controlled, parallel-arm study was completed in the USA in patients (n = 120) with persistent asthma. This trial assessed the safety, pharmacokinetics, and efficacy of anrukinzumab administered at three dose levels. Results showed that anrukinzumab did not meet the clinical efficacy endpoint, and its development was subsequently terminated in May 2009. IMA-026, another anti−IL-13 monoclonal antibody from the same company, was evaluated in phase I studies, and was progressed to phase II trials in 2009.
Kariyawasam et al. [62] studied the effects of the anti-IL-13 monoclonal antibody, QAX576, on inflammatory responses following nasal allergen challenges in patients with seasonal allergic rhinitis. This was a parallel-group, double-blind, placebo-controlled study (n = 16), with an open-label phase (n = 5) of topical nasal corticosteroids being administered. Subjects received intranasal timothy grass pollen (Phleum P5 allergen), and serial samples were collected using the synthetic absorptive matrix Accuwik® Ultra Medium (Pall Corporation, Port Washington, NY, USA). The nasal allergen challenge was performed at screening and on days 5, 6, and 7 following infusion with QAX576 or placebo. QAX576 inhibited IL-13 levels and, to a lesser extent, eotaxin levels during the late phase following the nasal challenge, although no apparent effects were detected in the QAX576 group on nasal lavage eosinophil levels or nasal symptom scores. In conclusion, QAX576 has a specific pharmacodynamic target, causing profound inhibition of nasal IL-13 responses.
CAT-354 is a human monoclonal IgG4 antibody shown to neutralise IL-13 in the murine airway and in human lung mast cells [43]. Phase I studies in subjects with moderate asthma have demonstrated that CAT-354 administered intravenously has an acceptable safety and tolerability profile, with good bioavailability and linear pharmacokinetics [63]. CAT-354 entered phase II trials in 2005 [64]. Since then, additional phase I and II clinical trials have been initiated for asthma in Europe, Australia, and the USA [65, 66].
Targeting IL-4/IL-13 receptor complex
AIR645, a dual inhibitor of IL-4 and IL-13, is a 2′-O-methoxyethyl second-generation antisense drug targeting the mRNA encoding the IL-4Rα subunit. Results from a phase I study evaluating the safety, tolerability and pharmacokinetics of once-weekly treatment with inhaled AIR645 in healthy volunteers demonstrated no serious adverse events or dose-limiting toxicities following a single or multiple doses of the drug [67]. Prolonged local exposure was observed in the airways, with a half-life of ∼5 days in induced sputum, which was independent of the dose. Plasma AIR645 exposure was reproducibly detected only at the top high doses (30 mg in single-dose arm; 20 mg in multiple-dose arm) administered, suggesting very low systemic drug absorption.
Bioengineered versions (muteins) of IL-4 have also been developed, which act as IL-4/IL-13 antagonists by binding to the shared IL-4Rα subunit and preventing oligomerisation of the subunits [68, 69]. These muteins consist of two types of IL-4 variants, one in which the Tyr124 residue is replaced with Asp and another in which the Arg121 residue is replaced with Asp (Tyr124Asp/Arg121Asp) [68, 69]. A recombinant human IL-4 variant, pitrakinra (AER 001, AerovantTM), which inhibits both IL-4R and IL-13R, has entered phase II clinical trials for the treatment of asthma. Encouraging preliminary “proof of concept” data from a phase IIa trial have been reported from an antigen challenge study (n = 30) wherein a dry powder formulation of pitrakinra (60 mg, delivered via inhalation) reduced the severity of LAR by 72% [70]. A decrease in the forced expiratory nitric oxide level was also reported, suggesting a reduction of airway inflammation. Additionally, treatment with pitrakinra resulted in no safety concerns associated with the drug [71].
Two candidates for soluble IL-13R currently exist: IL-13Rα1 and IL-13Rα2. Because the affinity of IL-13 for IL-13Rα2 is >10-fold higher than that for IL-13Rα1, soluble IL-13Rα2 is potentially a more robust blocking agent than its counterpart [72]. AMG-317 is a fully human monoclonal antibody currently under investigation for its ability to bind IL-4Rα, which functionally blocks the action of IL-4 and IL-13. In a double-blind, placebo-controlled study, AMG-317 was administered as single i.v. (healthy adults, n = 29; asthmatic adults, n = 6) or s.c. doses (healthy adults, n = 24; asthmatic adults, n = 7) [73]. No serious adverse events were reported; injection site reactions, headache and myalgia did occur, but at similar rates between the active and placebo groups. Pharmacodynamic evaluation demonstrated inhibition of thymus and activation-regulated chemokine (TARC), eotaxin-3 and monocyte chemotactic protein (MCP-4) production. A 10 mg dose completely inhibited IL-4- and IL-13-induced MCP-4 production and IL-13-induced TARC production.
A phase II, randomised, double-blind, placebo-controlled, multiple-dose clinical trial was conducted to determine the safety and efficacy of AMG-317 (75 mg, 150 mg or 300 mg s.c.) in patients with moderate-to-severe asthma [74]. The primary objective was to evaluate the efficacy of AMG-317 compared with placebo, as measured by the change in Asthma Control Questionnaire (ACQ) symptom scores from baseline to week 12. In 2008, interim results showed evidence of biological activity; however, the clinical efficacy data did not meet the expectations set for the trial (table 2⇑) [59].
A chimeric human IL-13 protein (cintredekin besudotox) was developed by Puri et al. [75] at the US Food and Drug Administration. This fusion protein comprises human IL-13 and a mutated form of the Pseudomonas exotoxin [76]. Cintredekin besudotox must be internalised into the target cell before it can exert its cytotoxic effects [77]. NeoPharm has licensed cintredekin besudotox from the National Institutes of Health, the US Food and Drug Administration and the University of Michigan as a potential therapeutic agent for the treatment of idiopathic pulmonary fibrosis and asthma.
Targeting STAT-6
Following binding of IL-13 to the IL-13 type II receptor complex, signals are transduced mainly via JAKs to phosphorylate the tyrosine residues of signalling molecules, such as the STAT proteins [7, 78]. Phosphorylated STAT-6 dimers translocate to the nucleus and activate the transcription of genes, including CD23 and major histocompatibility complex class II in B-cells, IL-4 and IL-13 in T-cells, and eotaxin (chemokine (CC motif) ligand 11) in fibroblasts. In experimental models, targeted gene disruption of STAT-6 inhibited AHR, airway inflammation, eosinophil infiltration and fibrosis [79]. Importantly, this approach of abrogating IL-13 signalling will also affect the signalling associated with IL-4 because of the STAT-6 activation upon IL-4 binding to its receptor. Signalling via IL-4Rα/IL-13Rα1 is thought to occur via IL-4Rα because stimulation of the receptor complex by IL-4 and IL-13 results in activation of signalling intermediates characteristic of IL-4 responses, including phosphorylation of IL-4Rα, insulin receptor substrate 2 and JAK1 [80]. Several experimental approaches directed at inhibiting STAT-6 are under investigation, including small-molecule inhibitors, antisense therapy and small interfering RNA, RNA interference and dominant-negative peptides, some of which are discussed below.
YM-341619 hydrochloride, which suppresses IL-4-induced STAT-6-dependent reporter gene expression, was shown to inhibit the differentiation of mouse spleen T-cells into Th2 cells in vitro [50]. Orally administered YM-341619 reduced plasma IgE levels in dinitrophenol (DNP)–Ascaris-sensitised rats, but not IgG2a levels. YM-341619 suppressed IL-4 and IL-13 production in the splenocytes of these DNP–Ascaris-sensitised rats without augmenting IFN-γ production. YM-341619 also led to dose-dependent suppression of eosinophil accumulation in the lung of ovalbumin-sensitised rats and AHR induced by repeated exposure to ovalbumin. These results suggest that YM-341619 has the ability to suppress allergen-induced Th2 responses by selectively inhibiting the differentiation of CD4+ T-cells into the Th2 subset [50].
CONCLUSIONS
A number of biological compounds are currently being developed for the treatment of asthma. Compelling pre-clinical and emerging proof-of-concept data suggest that the IL-4/IL-13/STAT-6 pathway plays a key role in the pathogenesis of asthma by promoting AHR and lung inflammation. To date, no safety concerns have been associated with any of the compounds discussed in this review; however, further studies are needed to evaluate their efficacy, as well as their long-term safety. The next challenge would be to develop biological compounds according to specific phenotypes determined based on atopy, lung function, age of asthma onset and duration, sex, symptoms, medication use and healthcare utilisation. Until more information is available with regard to IL-4-, IL-13- or STAT-6-specific asthma pathology, special patient populations with uncontrolled severe atopic asthma who may be more likely to benefit from novel IL-4-, IL-13- or STAT-6-targeted asthma treatment will need to be identified for study in future clinical trials evaluating these compounds.
Statement of interest
C.K. Oh, G.P. Geba and N. Molfino are employees of MedImmune LLC, a wholly owned subsidiary of AstraZeneca that develops biologics for the treatment of lung disorders and produces anti-IL-13 monoclonal antibody; C.K. Oh also holds AstraZeneca stocks.
Provenance
Publication of this peer-reviewed article was supported by MedImmune, LLC, USA (article sponsor, European Respiratory Review issue 115).
Acknowledgments
We thank V. Sevastita, formerly with MedImmune, LLC, Gaithersburg, MD, USA, for her editing support and thoughtful review and comments on the manuscript, and M. Tallin (MedImmune, LLC) for assistance with the literature search and product information.
- Received November 24, 2009.
- Accepted January 6, 2010.
- © ERSJ Ltd
References









