



Role of endothelin in lung fibrosis
- D. Abraham
- D. Abraham, Division of Medicine, Research Dept of Inflammation, Centre for Rheumatology and Connective Tissue Diseases, Royal Free and University College Medical School, University College London, Royal Free Campus, Rowland Hill Street, Hampstead, London NW3 2PF, UK. Fax: 44 2077940432. E-mail: d.abraham{at}medsch.ucl.ac.uk
Abstract
Accumulating evidence suggests that idiopathic pulmonary fibrosis (IPF) results from lung injury primarily following an abnormal wound healing response towards epithelial cell damage. Thus, rather than resulting from chronic inflammation, as thought previously, the inflammatory response acts as a modifier of the fibrogenic response, which can also be influenced by the host’s genetic background and environmental triggers. Several key mediators, including endothelin (ET)-1, have been implicated in the fibrosis and scarring associated with IPF.
Elevated levels of ET-1 have been detected in bronchoalveolar lavage fluid and serum from patients with IPF and increased expression of ET-1 has been detected in small pulmonary blood vessels and macrophages. In vitro data show that ET-1 influences matrix production and degradation by promoting synthesis of collagen type I and III, inhibiting expression of matrix metalloproteinase-1 and promoting matrix remodelling. In addition, ET-1 promotes fibroblast differentiation to a myofibroblastic cell type, inducing the expression of proteins that contribute to a contractile phenotype including α-smooth muscle actin. Moreover, ET-1 has been shown to initiate alveolar epithelial cell transition into fibroblast-like cells, a process termed epithelial–mesenchymal transition, and thereby contribute to pulmonary fibrosis. Furthermore, recently reported data from the BUILD (Bosentan Use in Interstitial Lung Disease)-1 trial showed a trend towards a delay in time to disease progression or death following treatment with the endothelin receptor antagonist, bosentan, which was especially prominent in patients with biopsy-proven IPF. This adds to the evidence that ET-1 plays an important role in IPF.
The present article examines recent evidence for the role of endothelin-1 in pulmonary fibrosis, and particularly, in the control of the function and differentiation of fibroblasts and myofibroblasts.
- Extracellular matrix
- fibroblasts
- idiopathic pulmonary fibrosis
- myofibroblasts
Increasing evidence supports the notion that the pattern of fibrosis seen in idiopathic pulmonary fibrosis (IPF) arises not primarily as a result of chronic inflammation, but owing to an abnormal wound repair response. This occurs following sequential alveolar injury, which leads to the progressive development of foci of scarring in the lung tissue, destruction of alveolocapillary units and loss of lung function [1]. Fibrosis and scarring is a complex process involving altered extracellular matrix (ECM) deposition, owing to changes or abnormalities in the normal balance of synthesis and degradation of newly formed ECM by fibroblasts and other mesenchymal cell types, particularly myofibroblasts. A wide range of factors have been identified as key mediators in fibrosis; recent research suggests that one such mediator with a key role in this process in the lung is endothelin (ET).
The ET family consists of three highly vasoactive peptides (ET-1, -2 and -3) with homologous structures. The three isoforms bind to two ET receptors (ET receptor subtype A (ETA) and subtype B (ETB)), which are widely expressed in a number of tissues, including the kidney, liver, lung and skin, and are known to be abnormally expressed in human diseases characterised by defective vascular control. The main, and most widely studied, isoform of ET is ET-1, which is produced primarily by endothelial cells, but also mesenchymal cell types, including fibroblasts and smooth muscle cells. The production of ET-1 is carefully regulated, and can be controlled by a number of soluble factors, including thrombin, angiotensin II, epinephrine, cytokines and growth factors, as well as physico-chemical stimuli, such as shear stress and hypoxia [2].
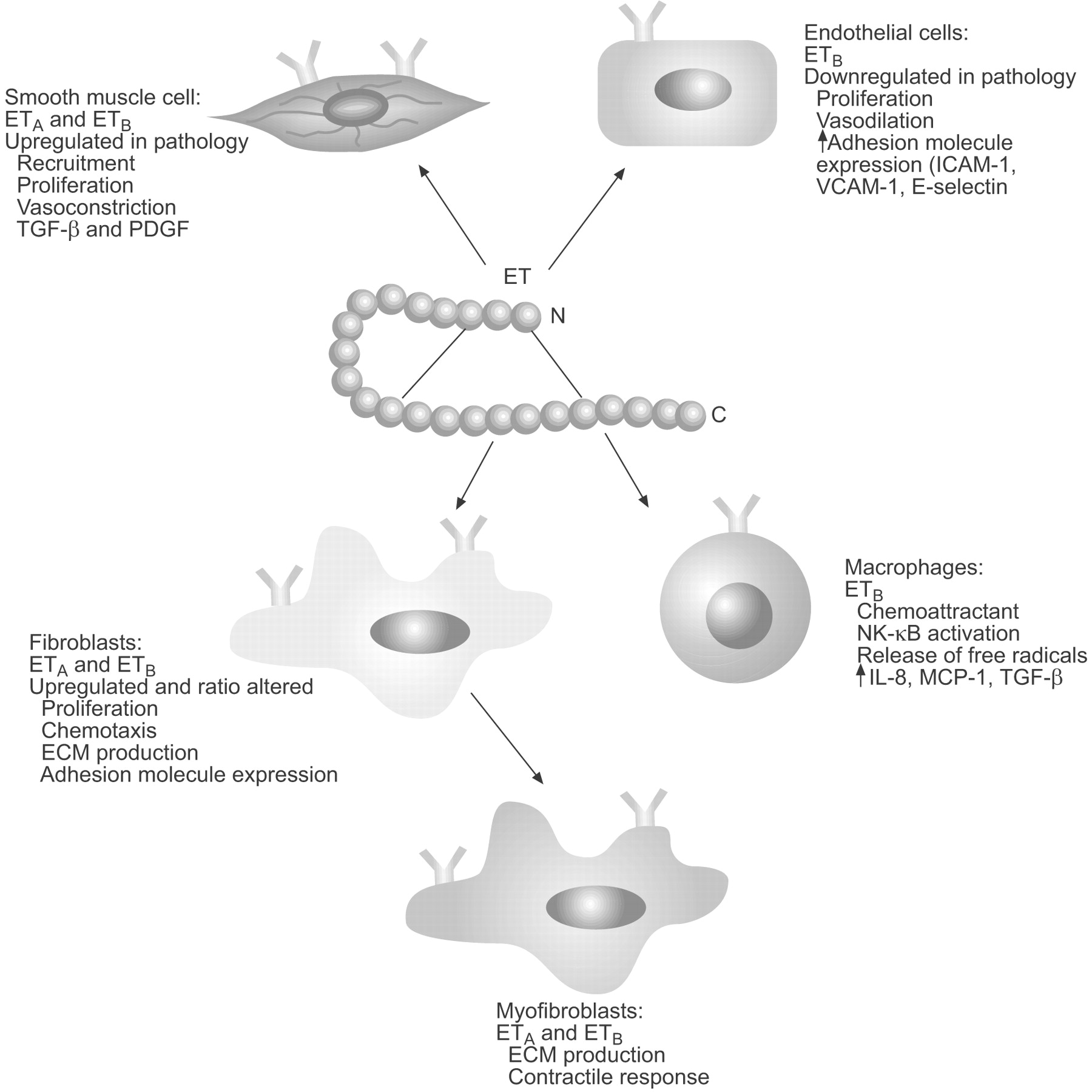
The ETA and ETB receptors have seven transmembrane domains and are coupled to G proteins. ETA receptors are predominantly expressed on mesenchymal cells (fibroblasts and smooth muscle cells) and mediate cell proliferation and vasoconstriction. ETB receptors act as the major clearance receptor for ET-1 and can participate in the release of nitric oxide. Sometimes alone, but primarily in combination with ETA receptors, ETB receptors can also mediate multiple detrimental effects in disease states, including cellular hypertrophy, inflammation and fibrosis [3]. ET-1 acts in a paracrine and autocrine manner to mediate its broad range of biological activities, which include the control of vasoconstriction and vasodilation via its action on the endothelium and vascular smooth muscle cells, a chemotactic effect on inflammatory cells, such as monocytes and neutrophils, and, as will be discussed further in the present article, a pivotal role in the function and differentiation of fibroblasts and myofibroblasts (fig. 1⇓).
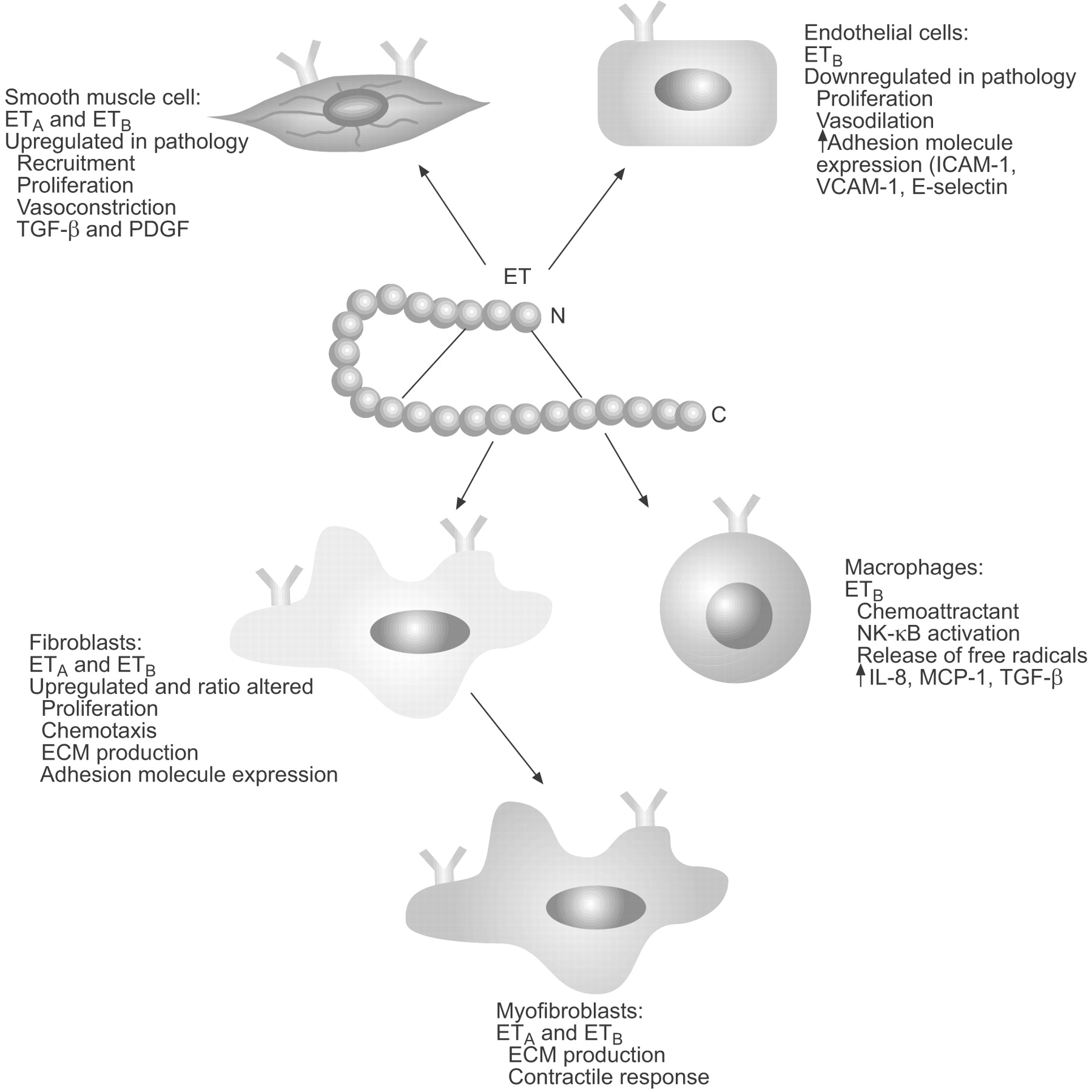
Biological activities of endothelin (ET)-1. ETA: ET receptor subtype A; ETB: ET receptor subtype B; ICAM: intercellular adhesion molecule; VCAM: vascular cell adhesion molecule; TGF: transforming growth factor; PDGF: platelet-derived growth factor; NK: nuclear factor; IL-8: interleukin-8; MCP-1: monocyte chemoattractant protein-1; ECM: extracellular matrix.
ET AND IPF
Elevated levels of ET-1 have been reported in bronchoalveolar lavage fluid and serum from patients with IPF [4, 5], and increased expression of ET-1 has been detected in small pulmonary blood vessels and macrophages [5]. In normal lungs, moderate expression of ET-1, its precursor big ET-1 and ET-converting enzyme (ECE) is localised to airway epithelium, pulmonary endothelium, and airway and vascular smooth muscle cells, indicating a role in normal lung physiology. However, in patients with IPF there is strong and extensive expression of ECE-1 in airway epithelium, proliferating type-II pneumocytes, and in endothelial and inflammatory cells [6]. Expression of ECE co-localised with big ET-1 (ET-1 precursor) and ET-1 immunostaining is significantly correlated with disease activity [6]. Saleh et al. [6] have also demonstrated that the proinflammatory cytokines interleukin-1 and tumour necrosis factor-α, which have been implicated in the pathogenesis of pulmonary fibrosis, increase ET-1 release and mRNA expression in human normal bronchial epithelial cells. Alveolar macrophages recovered from IPF patients show increased spontaneous secretion of ET-1 compared with cells from control subjects [7]. Interestingly, alveolar-associated fibroblasts from patients with IPF show increased expression of α-smooth muscle actin (α-SMA) and increased ET-1-induced contraction [8].
Further evidence for a role of ET-1 in pulmonary fibrosis comes from animal models. Mice transgenic for human pre-pro-ET-1 and transcriptional regulatory elements that overexpress ET-1 develop progressive pulmonary fibrosis with recruitment of predominantly CD4-positive inflammatory cells [9]. In addition, bleomycin-induced pulmonary fibrosis is associated with increased ET-1, ECE and big-ET-1 expression, as well as increased ET receptor expression [10–12]. Additionally, in studies utilising bleomycin-induced pulmonary injury in rats, collagen deposition was found to be reduced following treatment with the dual (ETA and ETB) ET receptor antagonist (ERA) bosentan, as measured by histology and tissue morphometry [10].
Taken together, these findings provide persuasive evidence for a role for ET-1 in the pathophysiology of pulmonary fibrosis in IPF and animal models. As discussed, ET mediates a wide range of biological activities which may be involved in fibrosis, among them the regulation of cell proliferation and differentiation, and the deposition and turnover of ECM.
ET IN FIBROBLAST FUNCTION AND DIFFERENTIATION
A complex series of events mediates the production of active ET-1 following gene transcription. The initially produced pre-pro-ET-1 peptides are progressively clipped by a host of converting enzymes to form first big-ET-1 and then mature ET-1. This final step is associated with a significant shift in biological activity, with ET-1 being around 500-fold more potent than the big-ET precursor. This complex mechanism between transcription and effect means there are a number of potential points at which the process might be inhibited. For example, the use of protease enzyme inhibitors to block ET-1 production, ET-induced signalling and transcription could be inhibited, or antagonists that block the binding of ET-1 to its receptors may be used [13].
As discussed, ET-1 is involved in a wide range of responses, depending on the receptor type and location, many of which have a potential role in pulmonary fibrosis. The activation of the ET receptor on mesenchymal cells leads to two distinct effects; a rapid, G-protein-mediated response via the inositol triphosphate signal transduction pathway, which leads to calcium release and smooth muscle contraction, and a slower differentiating and remodelling response. This slower response is mediated via a wide range of signalling cascades and transcription profiles that, in the fibroblast, lead to the activation of matrix-modifying genes and genes involved in fibroblast differentiation. It is this latter response which is of particular interest in pulmonary fibrosis [14].
The effects of ET-1 on collagen synthesis and fibroblast differentiation have been demonstrated in a number of in vitro systems. The addition of ET-1 to normal lung fibroblasts promotes synthesis of collagen types I and III, which can be inhibited by dual ERAs but not single ETA receptor antagonists [15]. This suggests that the mechanism is dependent upon both ETA and ETB receptors and implies that cross-talk, both between the receptors and downstream through various signalling pathways, is important for collagen production [15]. ET-1 can inhibit expression of matrix metalloproteinase (MMP)-1 (also known as collagenase) via ETA receptors, and the promotion of matrix remodelling was observed to be dependent upon ETA receptor expression [15]. As extracellular degradation of type-I collagen requires initial cleavage by MMP-1, the influence of ET-1 on interstitial MMP-1 expression could have substantial effects on matrix turnover.
More recently, the same group demonstrated that the overexpression of type-I collagen, fibronectin and connective tissue growth factor (CCN2; formerly known as CTGF) [16] in fibrotic fibroblasts can be reduced by treatment with the dual ET receptor antagonist bosentan [17]. Dual ET receptor antagonism also reduced the elevated adhesive property of fibrotic fibroblasts [17]. Conversely, inhibition with single antagonists for either the ETA or the ETB receptors was unable to efficiently reduce type-I collagen, fibronectin and CCN2 levels [17].
In addition to influencing matrix production and degradation, ET-1 promotes fibroblast differentiation to a myofibroblastic cell type, inducing the expression of proteins that contribute to a contractile phenotype, including α-SMA [18]. Shahar et al. [7] have shown that the expression of α-SMA in alveolar fibroblasts from IPF patients is significantly higher than in normal lung fibroblasts and can be increased in both cell types by ET-1. Shahar et al. [7] also demonstrated increased spontaneous secretion of ET-1 by alveolar macrophages from patients with IPF, and that neutralisation of ET-1 by monoclonal antibody decreased proliferation in fibroblasts cultured with alveolar macrophage supernatants. These results support a role for ET-1 in the modulation of α-SMA expression and proliferation in alveolar fibroblasts and, thus, in the pathogenesis of pulmonary fibrosis.
ET-1 also enhances the ability of lung fibroblasts to contract ECM in floating gels but not in fixed collagen gels [8, 19]. This presumably reflects the fact that ET-1 promotes matrix contraction via inducing myofibroblast formation, and does not directly promote stress fibre-mediated contraction by mechanical means [18]. This process can be blocked using ERAs [15]. Interestingly, relative to normal pulmonary fibroblasts, fibroblasts cultured from scars of patients with scleroderma show enhanced ET-1 expression and binding. These fibroblasts show an increased ability to contract a collagen matrix and an elevated expression of procontractile proteins, including α-SMA, which can be reduced using ERAs, implying that blocking ET-1 might be beneficial in reducing scar formation in pulmonary fibrosis (fig. 2⇓) [18].

Normal lung fibroblasts treated with or without endothelin (ET)-1 and bosentan for 24 h and subjected to immunofluorescence analysis with a mouse anti-α-smooth muscle actin (α-SMA) antibody and a fluorescein isothiocyanate-conjugated anti-mouse antibody. ET-1 elevated levels of α-SMA stress fibres in normal lung fibroblasts, and α-SMA levels were reduced following bosentan treatment. a) Treated with neither ET-1 nor bosentan; b) treated with ET-1 but not bosentan; c) treated with both ET-1 and bosentan. Reproduced from [18] with permission from the publisher.
ET AND TRANSFORMING GROWTH FACTOR-β
Along with ET-1, two of the other most potent profibrotic growth factors implicated in the development of fibrosis in a number of diseases, including pulmonary fibrosis, are transforming growth factor (TGF)-β and CCN2. As discussed, ET-1 promotes fibroblast-to-myofibroblast differentiation. It also interacts with other profibrotic cytokines, including TGF-β, a key component in the induction and progression of fibrosis, and is a potent stimulator of CCN2. In fact, CCN2 is a common target for both TGF-β and ET-1 [20]. Animal studies point to the existence of a cytokine hierarchy and interplay between ET-1, TGF-β and CCN2, which involves induction of ET-1 by TGF-β, induction of CCN2 by both ET-1 and TGF-β, and potential mediation of the effects of TGF-β and ET-1 on ECM by CCN2 (fig. 3⇓) [21].

Schematic diagram of the hierarchy and interplay between endothelin (ET)-1, transforming growth factor (TGF)-β and connective tissue growth factor (CCN2; formerly known as CTGF). NF: nuclear factor. Reproduced from [21] with permission from the publisher.
Studies have demonstrated that TGF-β induces ET-1 expression via a Smad-independent, c-Jun N-terminal kinase (JNK)-dependent mechanism [22, 23]. This mechanism has been further defined in a recent study by Shi-Wen et al. [24], which demonstrated that TGF-β induces ET-1 in normal and fibrotic lung fibroblasts in a Smad-independent ALK5 (TGF-β type-I receptor)/JNK/Ap-1-dependent fashion. In addition, ET-1 was found to induce JNK through TAK1. Constitutive JNK activation displayed by fibrotic lung fibroblasts can be reduced by dual ET receptor inhibition, providing evidence of an autocrine ET loop. This implies that ET-1 and TGF-β are likely to cooperate in the pathogenesis of pulmonary fibrosis, with increased JNK activation in fibrotic lung fibroblasts contributing to the persistence of the myofibroblast phenotype in pulmonary fibrosis by promoting an autocrine ET-1 loop (fig. 4⇓) [24]. The same group have also demonstrated that the ability of TGF-β to induce the expression of profibrotic genes, including type-I collagen, fibronectin and CCN2 in human lung fibroblasts, and to contract a collagen gel matrix, is dependent on ET-1 [25].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Endothelin (ET)-1 signalling pathways resulting in enhanced production of profibrotic proteins and extracellular matrix (ECM) production, remodelling and contraction. TGF: transforming growth factor; ALK5: TGF-β type-I receptor; JNK: c-Jun N-terminal kinase; AP: activator protein; SMA: smooth muscle actin. Reproduced and modified from [24] with permission from the publisher.
As well as the induction of profibrotic genes in lung fibroblasts, it has been postulated that TGF-β plays an important role in the pathogenesis of IPF and other forms of pulmonary fibrosis via its role in the epithelial–mesenchymal transition (EMT) of alveolar epithelial cells (AECs) [26]. There is also recent evidence of a role for ET-1 in this process: ET-1 has been shown to be produced at physiologically relevant levels by primary AECs and to be secreted preferentially toward the basolateral surface [27]. Furthermore, AECs express high levels of ETA receptors which suggests an autocrine or paracrine function for alveolar ET-1. Activation of ETA on AECs by ET-1 has been shown to induce EMT [27]. In addition, TGF-β synthesis is increased by ET-1 and ET-1-induced EMT is attenuated by a TGF-β-neutralising antibody, showing that ET-1 is an important mediator of EMT in AECs, possibly acting through ETA-mediated TGF-β production [27]. Overall, these studies demonstrate that ET-1 contributes to the profibrotic phenotype of fibrotic lung fibroblasts and, specifically, to the ability of TGF-β to promote this phenotype. This supports the notion that ET receptor antagonism may be beneficial in controlling fibrogenic responses in lung fibroblasts and, thereby, in combating fibrotic disease.
OTHER SOURCES OF FIBROBLAST AND MYOFIBROBLAST-LIKE CELLS
Data suggest that ET-1 plays an important role in a number of processes that may contribute to pulmonary fibrosis, a key aspect of which is myofibroblastic differentiation. As discussed, ET-1 is involved in driving the differentiation of activated resident fibroblasts to myofibroblasts, and there is also evidence to support the notion that ET-1 plays a role in EMT, providing an additional source of myofibroblastic cells from the airway epithelium [28, 29]. There is also increasing experimental evidence to suggest that endothelial-to-mesenchymal transition may play an important role during cardiovascular development and in human cardiopulmonary diseases characterised by vascular pathologies [30–32]. At certain stages of development, this process appears to be reversible and may, therefore, represent another target for therapeutic intervention. However, further research is necessary before the full contribution of endothelial-to-mesenchymal transition to pulmonary fibrosis is elucidated. There are also a number of other potential progenitor cells which may produce ECM components and/or undergo myofibroblastic differentiation, and ET may also have a role in these processes. For example, ET-1 is a potent chemotactic factor for circulating fibrocytes, cells which are known to participate in wound repair as well as in granuloma formation, antigen presentation and various fibrosing disorders [33]. Fibrocytes producing matrix proteins such as collagens I and III participate in the remodelling response by secreting MMPs and are thought to represent a systemic source of myofibroblasts in fibrotic lesions. Interestingly, in the bleomycin model, circulating mononuclear fibrocyte accumulation in the lung has been shown to contribute to the pathogenesis of pulmonary fibrosis [34].
CONCLUSIONS
The role of endothelin-1 in the development of pulmonary fibrosis is clearly complex, involving a number of progenitor cells and interaction of a range of key mediators in both paracrine and autocrine systems. Experimental evidence provides support for the role of endothelin-1 in myofibroblast formation and function, and also indicates a role for endothelin receptor antagonism in the moderation of this process. To date, such evidence largely exists in in vitro systems, and the relevance of any effect of endothelin receptor antagonists on myofibroblast formation and function in animal models and the clinical situation remains to be fully clarified. However, recently reported data from the BUILD (Bosentan Use in Interstitial Lung Disease)-1 trial of the dual endothelin receptor antagonist bosentan, which showed a trend towards a delay in time to disease progression or death that was especially prominent in patients with biopsy-proven idiopathic pulmonary fibrosis, adds to the evidence that endothelin-1 plays an important role in this fatal disease. The challenge for the future is to fully appreciate the role of endothelin-1 in pulmonary fibrosis at the cellular and molecular level.
Statement of interest
D. Abraham has received speaker fees and reimbursment of travel expenses from Actelion Biopharmaceutical and Encysive Pharmaceuticals.
- © ERSJ Ltd