





Abstract
Idiopathic pulmonary fibrosis (IPF), the most common of the interstitial pneumonias, is a progressive, life-limiting disease for which there are no truly effective therapies. In patients with biopsy-confirmed IPF, median survival is still <3 yrs. Although potent immunosuppressive therapy has underpinned the treatment of IPF in recent years and remains the standard of care, there is little quality evidence to support the efficacy and safety of traditional therapeutic strategies.
This has spurred the search for new treatments for IPF and has led to a series of clinical trials of new therapies, seven of which are reviewed herein. They include the Bosentan Use in Interstitial Lung Disease (BUILD)-1 trial, the results of which are discussed in detail, the European Idiopathic Pulmonary Fibrosis International Group Exploring N-acetylcysteine 1 Annual (IFIGENIA) trial, the interferon gamma (GIPF-001) trial and the INSPIRE trial, as well as trials of anticoagulant therapy, pirfenidone and etanercept.
Treatment trials in IPF are hindered by difficulties in achieving a secure diagnosis of IPF and the lack of validated outcome measures that represent either improvement or progression of disease. These and other limitations are discussed in the present article, as well as how some of these problems might be addressed in future trials.
Although few of the seven studies met their primary end-points, marginal trends either on primary end-points or statistically significant trends on exploratory end-points were a recurrent theme in most trials. In the BUILD-1 trial, for example, a trend in favour of bosentan was observed on time-to-disease progression or death.
Idiopathic pulmonary fibrosis (IPF) is a rare, progressive diffuse parenchymal lung disease and the most common of the interstitial pneumonias [1]. It is characterised histologically by a pattern of usual interstitial pneumonia [1].
Patients with IPF typically present with symptoms of nonproductive cough and progressive dyspnoea. IPF can have a variable course with some patients experiencing a progressive deterioration in lung function, while others develop acute exacerbations leading to rapid respiratory failure and early death [2]. Prognosis remains poor, especially among the subgroup of patients who develop acute exacerbations [2].
Until relatively recently, IPF was considered a chronic inflammatory disease, with treatment focused primarily on addressing inflammation through the use of potent immunosuppressive drugs, such as prednisone, azathioprine and cyclophosphamide. In fact, immunosuppressive therapy has been considered the standard of care for IPF by organisations such as the American Thoracic Society [3], although it was acknowledged in the international consensus statement that the evidence on which this strategy was based was limited and, indeed, some of the data contradictory [4]. However, as more has been discovered about the pathophysiological processes underlying IPF, new therapeutic approaches have been explored and controlled clinical trials conducted on these new therapies. For example, there is accumulating evidence that endothelin (ET)-1, a potent vasoconstrictor, plays an important role in the aetiology of IPF and that drugs that target ET-1 may be of benefit in patients with IPF [5, 6].
The present article summarises seven controlled clinical trials of new drug therapies for the treatment of IPF and discusses what lessons can be learnt from these trials, before examining in detail the outcome of the Bosentan Use in Interstitial Lung Disease (BUILD)-1 trial.
OVERVIEW OF CLINICAL TRIALS IN IPF
Clinical trials completed over the past 2–3 yrs have embraced a number of new therapeutic approaches to IPF and include, in addition to the BUILD-1 trial [6], the European Idiopathic Pulmonary Fibrosis International Group Exploring N-acetylcysteine 1 Annual (IFIGENIA) trial [7] and the interferon (IFN) gamma GIPF-001 trial [8], as well as trials on anticoagulant therapy [9], pirfenidone [10] and etanercept (table 1⇓) [11]. In addition, the INSPIRE trial, which was stopped early, also provides valuable information about the use of IFN-γ in IPF [12, 13].
Summary of seven treatment trials in idiopathic pulmonary fibrosis(IPF)
In the IFIGENIA trial, a multinational, double-blind, randomised, placebo-controlled trial, 182 patients with IPF were randomised to receive N-acetylcysteine (NAC) plus prednisone and azathioprine or placebo plus the same dosages of prednisone and azathioprine over a 12-month period [7]. Changes from baseline in forced vital capacity (FVC) and diffusing capacity of the lung for carbon monoxide (DL,CO) were primary end-points, while secondary end-points included clinical, radiological and physiological score, high-resolution computed tomography (HRCT) findings, quality of life (QoL) and survival. There was a significant slowing in the rate of decline from baseline in both FVC and DL,CO in the NAC treatment arm and these findings suggest that the addition of NAC to low-dose prednisone and azathioprine may help to preserve pulmonary function in patients with IPF. However, the delay in disease progression did not result in increased survival as there was no significant difference between treatment arms with respect to survival at 12 months.
The GIPF-001 trial was a randomised, double-blind, placebo-controlled study in 330 patients with IPF, which assessed the effect of IFN-γ on progression-free survival, defined as the time-to-disease progression or death, as well as pulmonary function and QoL [8]. Over a median of 58 weeks there was a trend towards improved survival (primary end-point) in the IFN-γ cohort, although the difference failed to reach statistical significance. However, among patients with less severe lung impairment at baseline, the difference in survival between IFN-γ and placebo was significant (4 and 12% mortality, respectively; p = 0.04). No significant treatment effect was observed on measures of lung function, gas exchange or QoL. The effect of IFN-γ in patients with IPF was also assessed in the multicenter, double-blind, randomised INSPIRE study with survival time as the primary end-point [12]. However, this study was stopped prematurely following a planned interim analysis. The results showed that overall survival had crossed the predefined stopping boundary for lack of benefit, although among the 826 randomised patients there was not a statistically significant difference between treatment groups in overall mortality (14.5% in the IFN-γ group compared with 12.7% in the placebo group).
The potential benefits of anticoagulant therapy on survival in IPF were evaluated by Kubo et al. [9], in a study of 56 patients in which the effects of prednisolone alone were compared with prednisolone plus an anticoagulant (warfarin or low molecular weight heparin). A significant difference was observed between the survival curves of the anticoagulant and non-anticoagulant groups, with a mortality hazard ratio of 2.9 (p = 0.04). Mortality associated with acute exacerbations of IPF in the anticoagulant group was also significantly reduced compared with that in the non-anticoagulant group (18 versus 71%, respectively; p = 0.008). No significant difference was evident between groups with respect to hospitalisation-free periods.
In the randomised, double-blind, placebo-controlled study to investigate the effects of pirfenidone in IPF, 107 patients with mild-to-moderate disease received either pirfenidone or placebo for 12 months [10]. Although the study did not meet its primary end-point of improvement on lowest arterial oxygen saturation measured by pulse oximetry (Sp,O2) with exercise, more patients receiving pirfenidone experienced an improvement in the minimum Sp,O2during the 6-min exercise test compared with placebo, while fewer patients experienced a decline in the minimum Sp,O2. There was also a significant difference between pirfenidone and placebo with respect to secondary end-points of change in vital capacity and acute exacerbations. In fact, this study was halted prematurely due to an increase in acute exacerbations in the placebo arm. In the 6 months following randomisation, five acute exacerbations occurred among 35 patients in the placebo arm, compared with none in the pirfenidone treatment group.
The trial of etanercept was a prospective, randomised, placebo-controlled study in 87 IPF patients, in which patients were assessed after 12-months treatment with respect to change from baseline FVC (% predicted), DL,CO (% pred), and alveolar–arterial oxygen gradient (A–a) gradient [11]. Again, this trial failed to meet its primary end-point. However, in the etanercept treatment arm there was a trend toward reduced disease progression by several measures in these patients with progressive IPF.
LIMITATIONS OF CLINICAL TRIALS IN IPF
Although the choice of primary end-point differed between the trials summarised previously, it was notable that with few exceptions, such as IFIGENIA, the trials failed to meet their primary end-points. The anticoagulant study by Kubo et al. [9] also demonstrated significant treatment effects but the study design was seriously flawed. To start, patients were not blinded to treatment and an intention-to-treat analysis was not used, which could have skewed the results. Moreover, over a quarter of patients in the anticoagulant group withdrew before treatment was initiated.
Although few studies met their primary end-points, marginal trends either on primary end-points or statistically significant trends on exploratory end-points favouring the investigational treatment under study were a recurrent theme throughout all of the studies. In the BUILD-1 trial, for example, there was a trend to delayed time-to-disease progression or death in the bosentan treatment group, while in the GIPF-001 trial there was a significant improvement in survival among the less severely impaired patients treated with IFN-γ. These findings suggest that either a treatment broadly lacks efficacy but achieves a low average effect or, alternatively, is very selective but achieves important efficacy in selected subgroups. The results from these trials also raise important questions about both the selection of patients for inclusion in IPF treatment trials and the selection of appropriate outcome measures.
Selection bias is an acknowledged and complex problem in IPF trials making it difficult to draw useful comparisons between studies. Among the trials, BUILD-1 was unique in that it linked the treatment effect to baseline morphological characteristics. However, it is unclear whether the BUILD-1 findings were truly unique since HRCT scan results were not handled in a comparable way in other studies where there was some evidence of a treatment effect. Apart from the GIPF-001 trial, where a comparable 65% of patients also underwent surgical lung biopsy, the proportion of patients biopsied in the other trials was much lower than in BUILD-1; ranging from 49% in IFIGENIA to <20% in the studies by Azuma et al. [10] and Kubo et al. [9]. The observed severity of disease also points towards differences between studies, as the patient populations enrolled in the two Japanese trials tended to exhibit milder disease.
Inconsistencies in patterns of disease progression were also evident between the six studies. In an analysis of data from the placebo treatment arm of the GIPF-001 trial, Martinez et al. [2] noted a surprisingly high prevalence of acute exacerbations compared with that seen in usual clinical practice. In contrast, there was little evidence of the more usual rapid typical progression, in which lung function deteriorates over a 6–12-month period. This suggests that such IPF patients may have declined to participate in the study because they perceived their disease to be more rapidly progressive. This is an interesting finding in the context of the IFIGENIA study, where the placebo arm constituted “best current practice”. Here there was a much higher prevalence of typical, more rapidly progressive IPF, reflected by significant change in FVC in 1 yr, and a much lower prevalence of acute exacerbations. Clearly bias can have important effects, both between studies and in distancing study populations from routine clinical practice and, as such, should be defined pro-actively.
Three issues are important when considering the choice of outcome measure in IPF trials: 1) end-point reproducibility; 2) the process actually being measured by a given end-point; and 3) how should the signal best be expressed and analysed. In the GIPF001 (IFN-γ) study, change in the A–a gradient was a primary pulmonary function test end-point. However, the change threshold of 5 mm occurs frequently from measurement variation and, moreover, does not predict mortality [12, 14]. In this case, it was simply a reproducibility of signal issue, where there was a high noise to signal ratio. Exercise capacity measured by 6-min walking distance (6MWD) is an established end-point in pulmonary arterial hypertension (PAH) trials and, in principle, should be an excellent end-point in IPF longitudinal studies as well. It was used as an end-point in both the BUILD-1 and etanercept trials, but in neither trial was this end-point met. There is evidence from clinical experience to suggest that there are some IPF patients who learn to change their pattern of breathing with restrictive lung function, so that over a period of time with the same disease severity, they walk with less respiratory work and their walk distance increases. Conversely, there are patients who become depressed, decompensate and the 6MWD declines while severity remains unchanged. Thus, it can be argued that in IPF the 6MWD is a “catch-all” end-point, catching patient adaptation to disease as well as change in severity.
The next important issue in selecting end-points is to define what the trial is designed to capture, e.g. typical decline, acute exacerbation or progressive pulmonary vascular disease, and how closely it should mirror usual clinical practice. An all encompassing end-point might be a combination of progression-free survival using FVC and death covering both typical decline and acute exacerbations. However, if there is a concern about selection bias, then separate end-points might include serial FVC, and separately hospital admissions and death. Finally, there is the issue of how the signal should best be expressed and analysed. The conventional wisdom is that continuous change is superior and more sensitive, and that is certainly true for a unimodal disease, where progression can be described to 2 sd. However, for a disease such as IPF where there are striking differences between subgroups, and evidence of differential treatment effects, categorical analysis is often more sensitive. On the assumption that subgroup analysis can be justified, it is logical to compare the outcome of continuous and categorical analyses to see if they are giving different messages. This is nicely illustrated in the IFIGENIA study, where, it can be argued, the NAC-treated group had a bimodal pattern of disease progression, with little average decline but major decline in an important subgroup, suggesting a differential treatment effect. In BUILD-1, there was very little difference in continuous signal but again, categorical trends suggested a subgroup treatment effect. Therefore, a strong case can be made for being more pro-active in the design of trial protocols, detailing both the intention to perform subgroup analyses, and the intention to compare continuous and categorical signals.
Security of diagnosis is also of fundamental importance in the conduct of IPF clinical trials and can help to address some of the problems of selection bias. In this regard, the key question is whether or not to have a central HRCT and histological review before patient enrolment into a trial. The advantage of a central review is that it restricts the disease to an epithelial/fibrotic disorder, thus any observed treatment effects are more specific to IPF; failure to perform central review results in the variable addition of patients with an inflammatory/fibrotic pathogenesis. While any observed treatment effects are more applicable to usual clinical practice, it is much harder to attach them specifically to an effect on IPF. There is an unanswerable case for central review to improve diagnostic accuracy in IPF trials. First, on the basis of available evidence, subgroup analysis is crucial. Secondly, IPF is a difficult disease to diagnose and it is important to establish that it can be treated when diagnosed accurately before analyses based upon “intention to diagnose”, based on routine diagnostic practice, can be addressed. Thirdly, an intention to diagnose design pre-supposes that the community standard of diagnosis of IPF will remain precisely the same over the next 5–10 yrs, which is highly unlikely to be the case. Already, expert review will divert diagnosis away from IPF in ≤25% of cases. These findings suggest that central diagnostic review before enrolment in a clinical trial ought to be strongly considered during the present phase of IPF treatment research.
REVIEW OF BUILD-1 DATA
As the trial summary in table 1⇑ shows, BUILD-1 was a large, multinational, randomised, double-blind, placebo-controlled trial of bosentan in a well-defined population of 158 patients with IPF [6]. Notable exclusions were patients with evidence of PAH and those with disease duration of >3 yrs, since patients with a longer duration of disease might be a different phenotype given that median survival for IPF is ≤3 yrs. To exclude patients with end-stage disease, only those with an FVC of >50% and a DL,CO at baseline of >30% were included. Additional inclusion criteria were a 6MWD of 150–500 m and an Sp,O2of >80%.
Patients received 12 months treatment with either bosentan, titrated to a goal of 125 mg b.i.d, or matching placebo. Of the 158 randomised patients, a total of 49 prematurely discontinued (25 bosentan and 24 placebo), primarily due to adverse events including worsening in IPF. While bosentan-treated patients had a higher incidence of hepatic aminotransferase elevations compared with placebo, they had a lower incidence of respiratory adverse events, most notably cough (17.6 versus 27.4%), exacerbation of dyspnoea (13.5 versus 19.0%) and worsening of IPF (16.2 versus 23.8%). At the end of the 12-month treatment period, analyses of the 109 patients who completed the study (49 bosentan and 60 placebo) showed no significant difference between treatment arms with respect to the primary efficacy end-point of 6MWD. However, a trend in favour of bosentan was observed in the secondary end-point of time to death or disease progression, defined as worsening pulmonary function tests or acute decompensation (fig. 1⇓, table 2⇓). A post hoc analysis of these data showed that in the subset of patients who underwent surgical lung biopsy because of atypical HRCT scans, the difference between bosentan and placebo reached statistical significance (fig. 1⇓, table 2⇓). Thus, in this subset of patients with biopsy-proven IPF, treatment with bosentan appears to be associated with slowing of disease progression.

Time to disease progression or death in a) the whole Bosentan Use in Interstitial Lung Disease (BUILD)-1 study population and b) the subpopulation with biopsy-proven idiopathic pulmonary fibrosis. ·········: treatment with bosentan, ––––: placebo. a) Hazard ratio (95% confidence interval): 0.613 (0.328–1.144); p = 0.119. b) Hazard ratio (95% confidence interval): 0.315 (0.126–0.789); p = 0.009.
Time to disease progression or death in the Bosentan Use in Interstitial Lung Disease-1 study
Additional post hoc analyses of the effect of bosentan on exercise-induced Sp,O2 in a group of patients who desaturated during the baseline 6-min walk test (≥4% decrease in Sp,O2 during exercise) showed that, while almost none of the placebo group showed evidence of improvement in exercise-induced desaturation, improved gas exchange with activity was seen in a number of bosentan-treated patients (fig. 2⇓). Furthermore, the difference between treatments was statistically significant. Overall, although the development of new desaturation did not differ between treatment arms in those patients who did not desaturate at baseline, in those who did, Sp,O2 improvement occurred in significantly more bosentan-treated patients than among placebo recipients.

Improvement in oxygen saturation in patients who desaturated during baseline assessment of 6-min walking distance in the Bosentan Use in Interstitial Lung Disease (BUILD)-1 study. In total, one patient out 57 in the placebo group and seven out of the 56 receiving bosentan showed a ≥4% improvement. Sp,O2: arterial oxygen saturation measured by pulse oximetry. #: p = 0.032.
Dyspnoea is a cardinal feature of IPF and drives most of the QoL issues for patients with IPF. Baseline values for both the Baseline Dyspnoea Index and the Transitional Dyspnoea Index showed significant and comparable impairment in patients randomised to bosentan and placebo in BUILD-1. Assessments following 6 and 12 months of treatment showed, not unexpectedly, continued deterioration in dyspnoea among a majority of placebo-treated patients; they became more breathless at 6 months and even more breathless at 12 months. Although the same pattern was seen in the bosentan-treated patients, the rate of decline was significantly less at 6 months and less at 12 months, though statistical significance was not maintained at 1 yr (fig. 3⇓). Bosentan had a similar effect in the subpopulation of patients with biopsy-proven IPF. These findings suggest that beyond 6 months, progression of disease may outstrip drug efficacy.
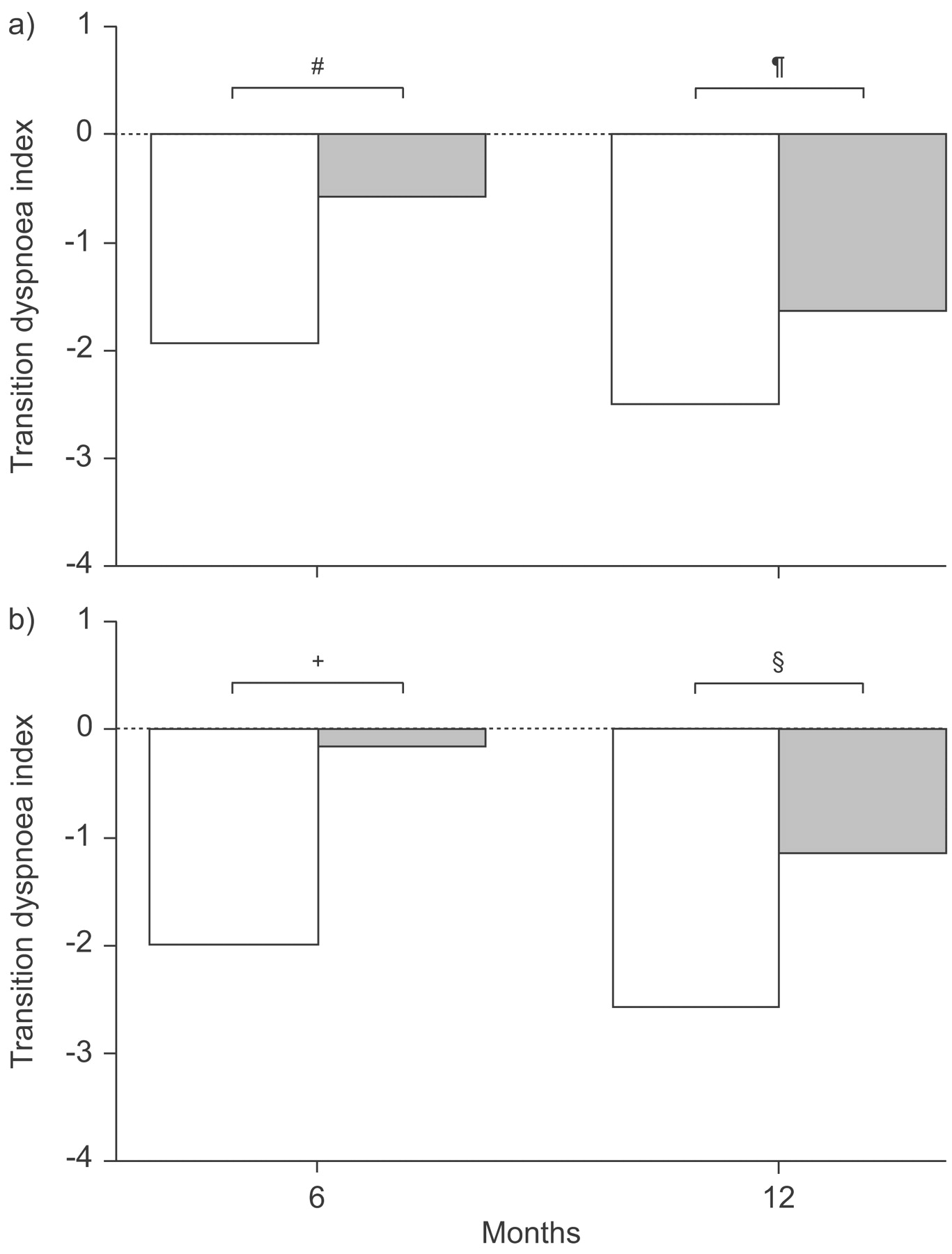
Effect of study medication on transition dyspnoea index at 6 and 12 months in a) the whole Bosentan Use in Interstitial Lung Disease (BUILD)-1 study population and b) the subpopulation with biopsy-proven idiopathic pulmonary fibrosis. □: bosentan (n = 83); ▓: placebo (n = 71). #: p = 0.016; ¶: p = 0.292; +: p = 0.015; §: p = 0.159.
In parallel with the findings on dyspnoea, treatment with bosentan was found to improve overall health-related QoL, as measured by the Medical Outcomes Study Short-Form 36-item instrument, as well as respiratory-specific QoL, as measured by the St George's Respiratory Questionnaire. Once again, the difference between treatments was significant at 6 months; however, while the trend was maintained the difference lacked statistical significance at 12 months (fig. 4⇓). In the biopsy-proven IPF subpopulation, the treatment effect on QoL was more pronounced than for the study population as a whole. Together with the results on time-to-disease progression or death, these findings suggest that bosentan may be of particular benefit in this patient subgroup.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of study medication on respiratory-specific quality of life in the Bosentan Use in Interstitial Lung Disease (BUILD)-1 study. □: bosentan (n = 80); ▓: placebo (n = 66). #: p = 0.009; ¶: p = 0.147.
CONCLUSIONS
Conventional approaches to the treatment of IPF have been largely empirical, predicated on the belief that IPF was a chronic inflammatory disease. With greater understanding of the pathophysiology of IPF, a more evidence-based approach to the treatment of IPF is evolving. Six recent clinical trials on potential new therapies for IPF highlight not only possible treatment effects but also problems inherent in the design of treatment trials in IPF patients. Selection bias is a key consideration in treatment studies, in which a large number of factors may be involved. These range from varying severity and disease duration at baseline to problems in obtaining a secure diagnosis of IPF prior to enrolment. To this end, central diagnostic review before enrolment in a clinical trial ought to be mandatory during the present phase of IPF treatment research. Selection bias needs to be confronted and understood and not simply swept under the carpet. Choice of outcome measures in IPF treatment trials is still an unresolved issue; end-points must distinguish between typical decline, acute exacerbations and progressive pulmonary vascular disease. Reproducibility is another key end-point issue; for different reasons, the A–a gradient and 6MWD are not ideal end-points in IPF trials. A search for subgroup effects is strongly warranted, especially when subgroup behaviour is evident from comparisons of continuous and categorical signal. This needs to be articulated prospectively in trial protocols.
Among several new therapies under investigation, there is evidence to suggest that treatment with NAC, bosentan and pirfenidone, may have clinical benefit in some, if not all, IPF patients. Although in the BUILD-1 trial, bosentan had no effect on the primary end-point of 6MWD, a trend in favour of bosentan was observed on time-to-disease progression or death. Post hoc analysis showed that this treatment effect was more pronounced in a predefined subpopulation with biopsy-proven IPF. Treatment with bosentan also resulted in measurable benefits in dyspnoea and QoL. Again, these effects were more pronounced in the subpopulation of patients with biopsy-proven IPF.
However, the negative results obtained following interferon-γ treatment in the INSPIRE trial highlight the need for large, well designed studies in patients with idiopathic pulmonary fibrosis before drawing firm conclusions about the benefits of a new therapeutic approach.
Statement of interest
A.U. Wells has received reimbursement as a speaker at a symposium organised by Actelion and has also received a grant for research from Actelion, including funding for a research fellow. A.U. Wells has received consultancy fees from Centricorp and Genzyme. K.K. Brown has served as a consultant member for IPF trials supported by Actelion. He has also received fees for attending a symposium, speaking, organising education and research.
- © ERSJ Ltd









