



Dual endothelin receptor antagonism: setting standards in PAH
- M. Humbert
- M. Humbert, Service de Pneumologie et Réanimation Respiratoire, Hôpital Antoine-Béclère, 157 rue de la Porte de Trivaux. Fax: 33 146303824. E-mail: marc.humbert{at}abc.aphp.fr
Abstract
Endothelin (ET) has emerged as a key mediator in the pathophysiology of pulmonary arterial hypertension (PAH). The effects of ET are mediated by its binding to two receptors on endothelial and pulmonary smooth muscle cells: ETA and ETB. Blockade of both these receptors with the oral dual ET receptor antagonist, bosentan, represents an attractive treatment option for these severely compromised patients.
The efficacy of bosentan in PAH has been demonstrated in randomised controlled trials in idiopathic PAH, and PAH associated with connective tissue diseases and congenital heart disease.
In addition, an open-label study has shown clinical and haemodynamic effects in PAH associated with HIV infection. In these trials, bosentan has been shown to improve haemodynamics, increase functional capacity, improve time to clinical worsening, which is a surrogate marker of survival, and to improve patients' quality of life; in longer-term studies, it has been shown to improve patient outcome.
The current article will present these key data from randomised controlled trials on bosentan as well as experiences from everyday clinical practice.
- Bosentan
- dual endothelin receptor antagonism
- pulmonary arterial hypertension
Increasing knowledge about the pathophysiology of pulmonary arterial hypertension (PAH) has led to the identification of endothelin (ET)-1 as a key mediator in the pathogenesis of this devastating condition [1]. ET-1 is a peptide, which mediates its effects by binding to two receptors on endothelial and pulmonary smooth muscle cells: ETA and ETB. Binding of ET-1 to these receptors is blocked by the dual ET receptor antagonist bosentan and there is significant preclinical evidence to suggest that this blockade inhibits the effects of ET-1 in the pathogenesis of PAH and other ET-1-mediated conditions [2, 3].
CLINICAL EVIDENCE IN SUPPORT OF BOSENTAN IN IDIOPATHIC PAH PATIENTS
Well-designed, placebo-controlled, randomised trials have generated a wealth of data to support the use of bosentan, not just in idiopathic PAH (iPAH) but also in PAH associated with conditions such as connective tissue diseases (PAH-CTD), congenital heart defects (PAH-CHD) and HIV infection.
The efficacy of bosentan was first demonstrated in a study by Channick et al. [4], which showed its efficacy in patients with iPAH or PAH-CTD. In that study, bosentan resulted in improved haemodynamics (increased cardiac index (CI), decreased mean pulmonary arterial pressure and decreased pulmonary vascular resistance (PVR); fig. 1⇓), as well as significant improvements in functional capacity as evidenced by improvements the 6-min walk distance (6MWD), Borg dyspnoea index and New York Heart Association/World Health Organization (WHO) functional class (NYHA FC) [4]. On the back of this, the larger Bosentan Randomized trial of Endothelin Antagonist THErapy (BREATHE)-1 trial was undertaken [5], which was a 16-week study in 213 patients with iPAH or PAH-CTD.

Bosentan improves haemodynamic parameters in pulmonary arterial hypertension patients (changes from baseline to week 12). Compared with placebo (n = 10), bosentan (n = 20) resulted in significant improvements in a) cardiac index (CI), b) mean pulmonary arterial pressure (P̄pa) and c) pulmonary vascular resistance (PVR) after 12 weeks. a) p<0.0001; b) p = 0.013; c) p = 0.0002.
As well as corroborating the symptomatic relief results of the smaller study of Channick et al. [4] by confirming the positive effect of bosentan on 6MWD (fig. 2⇓), BREATHE-1 resulted in a significant improvement in time to clinical worsening (TTCW; fig. 3⇓), a combined end-point measuring longer-term outcomes, not just a measure of exercise capacity and symptomatic relief, and defined as death, hospitalisation, premature discontinuation due to worsening PAH, start of prostanoid therapy, lung transplant or septostomy. It is clearly unethical to perform a placebo-controlled survival study in these patients, so evaluating end-points such as TTCW is useful as a surrogate marker of survival.

Bosentan improves exercise capacity in pulmonary arterial hypertension patients. At week 4 the dosage was changed from 62.5 mg to 125 mg or 250 mg b.i.d. After 16 weeks of treatment, the 6-min walk distance increased by 36 m in the bosentan group (combined doses of 125 mg or 250 mg b.i.d.), whereas a deterioration of 8 m occurred in the placebo group, a mean difference of 44 m. •: bosentan (n = 144); ▪: placebo (n = 69). p = 0.0002.

Bosentan delays time to clinical worsening. Compared with placebo, over the course of 28 weeks, bosentan significantly delayed time to clinical worsening. The difference between treatment groups was apparent as early as week 16 and was significant when the data from patients who continued into period 2 were censored at the week 16 assessment. ----: bosentan 125 or 250 mg b.i.d.; –––––: placebo. p<0.004 comparison between bosentan and placebo at week 16; p<0.01: comparison between bosentan and placebo at week 28.
Even longer-term effects of bosentan have been evaluated in open-label extensions of the two pivotal trials. In the open-label extension phase of the study by Channick et al. [4], 29 out of 32 patients received bosentan for an additional year (ex-placebo n = 8; ex-bosentan n = 21). Of the patients who had received bosentan in the double-blind phase of the trial, the improvement in 6MWD was maintained after 6 months of additional bosentan therapy (mean±sem: 60±11 m). In those who had previously received placebo, the 6MWD improved significantly compared with baseline (45±13 m). Long-term treatment with bosentan for >1 yr was associated with an improvement in haemodynamic parameters and NYHA FC.
The ultimate aim of therapy for any relentlessly progressive disease is to demonstrate a positive effect on survival. An open-label extension study of the patients enrolled in the study by Channick et al. [4] and BREATHE-1 compared observed long-term outcome up to 36 months, reported as Kaplan–Meier estimates, with predicted survival for each patient calculated using the validated National Institutes of Health (NIH) registry formula [6, 7]. The results of this study [8] demonstrated that bosentan had a positive effect on long-term outcome in patients with advanced PAH (table 1⇓).
Kaplan–Meier estimates and predicted survival using the validated National Institutes of Health registry formula [6]
EFFECT OF BOSENTAN IN A NONSELECTED CLINICAL POPULATION
Since the spectrum of patients in clinical trials is unlikely to be the same as that seen by clinicians in a “real world” situation, a recent retrospective analysis of 103 iPAH patients has been performed, which assessed the long-term outcome of patients in NYHA FC III (91 out of 103) or IV (12 out of 103) treated with first-line bosentan [9]. In their study, Provencher et al. [9] evaluated a number of end-points, including overall survival, survival without lung transplantation, acute right heart failure with hospitalisation and need of i.v. diuretics and/or dobutamine infusion, and need for i.v. prostacyclin therapy (whether it was administered or not). At the Antoine-Béclère Hospital (Clamart, France), the need for i.v. epoprostenol therapy was indicated when the patient was in NYHA FC IV on treatment or persisted in NYHA FC III after ≥4 months of treatment, together with at least one of the following: 1) a 6MWD <250 m; 2) a >10% decrease in 6MWD from the previous value in two tests performed ≥2 weeks apart; or 3) CI <2.2 L·min−1·m−2. Effects on 6MWD and haemodynamics were also recorded.
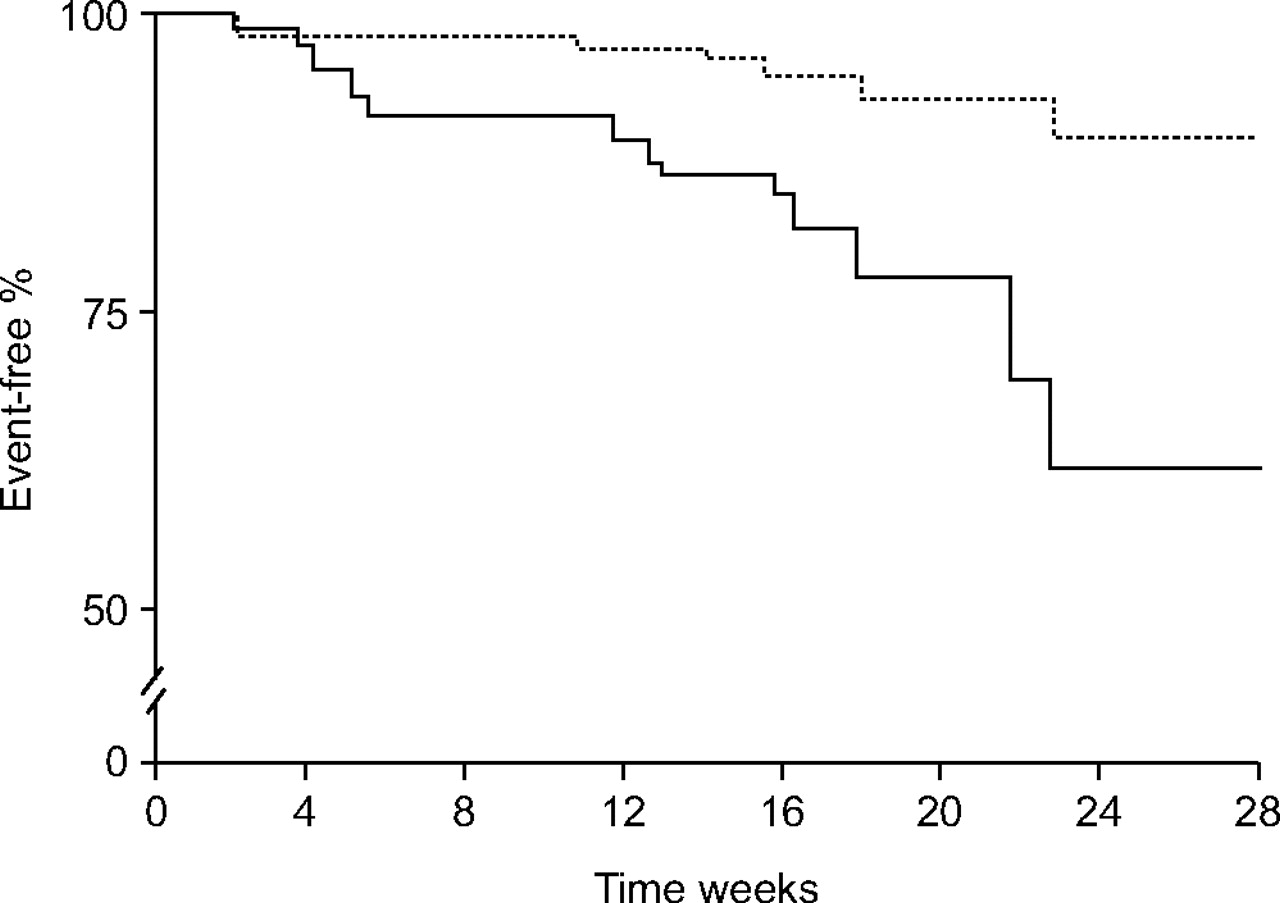
In this study [9], exercise capacity evaluated using the 6MWD increased from 322±15 m at baseline to 364±109 m at 4 months (p<0.001), which was in line with the results obtained in the placebo-controlled study of bosentan [5]. In accordance with this, haemodynamic parameters also improved. These positive changes in exercise capacity and haemodynamics persisted at 1 yr [9]. The overall survival estimate at 2 yrs was 89% compared with only 45% predicted by the equation derived from the NIH registry (fig. 4⇓) [6]. Although 44% of patients survived “event-free” at 2 yrs, this means that >50% were in need of some additional intervention, be it i.v. prostacyclin or lung transplantation.

Long-term outcome and event-free status in pulmonary arterial hypertension (PAH) patients treated with first-line bosentan. Kaplan–Meier estimates of survival and event-free status in 103 patients with idiopathic PAH treated with first-line bosentan therapy. Estimated survival rates (––––––) and estimates of event-free status (-----) are shown. Event-free status was defined as survival without lung transplantation, need for prostanoid therapy, or acute right heart failure requiring hospitalisation for i.v. diuretics and/or dobutamine infusion. For patients with more than one event, only the first event was used in the analysis. Reproduced and modified from [9] with permission from the publisher.
In line with earlier results, long-term outcome was directly related to 6MWD. Patients who were able to walk further at both baseline and 4 months survived for longer than those who could only walk shorter distances (fig. 5⇓). It is important to note that only patients who continue to respond should remain on monotherapy; those who initially respond and then remain stable or deteriorate should be considered for additional therapies added onto bosentan. Provencher et al. [9] reported a number of variables that were associated with long-term outcome, including 6MWD and right atrial pressure at baseline, and 6MWD and change in total pulmonary resistance (TPR) after 4 months of bosentan therapy (table 2⇓).

Long-term outcome in pulmonary arterial hypertension (PAH) patients according to baseline and 4-month 6-min walk distance (6MWD). a) Kaplan–Meier survival estimates in 103 patients with idiopathic (i)PAH grouped by the baseline 6MWD. Survival rates for patients with a baseline value >330 m (––––––) were 98, 98 and 91% at 1, 2 and 3 yrs, respectively, compared with 86, 79 and 66%, respectively, for patients with a baseline value <330 m (-----; p = 0.002 by Cox–Mantel log-rank test). b) Kaplan–Meier survival estimates in 99 patients with iPAH grouped by the 6MWD after 4 months of bosentan monotherapy. Survival rates for patients with a value >378 m after 4 months (———) were 100, 100 and 90% at 1, 2 and 3 yrs, respectively, compared with 85, 79 and 69%, respectively, for patients with a value <378 m (-----; p = 0.005 by Cox–Mantel log-rank test) after 4 months. Reproduced and modified from [9] with permission from the publisher.
Variable associated with survival
The results from this retrospective, real-life clinical patient population provide additional support to data derived from the analysis of iPAH patients treated with bosentan in the two placebo-controlled clinical studies and their extensions, in which bosentan improved long-term outcome.
PAH ASSOCIATED WITH OTHER AETIOLOGIES
Connective tissue diseases
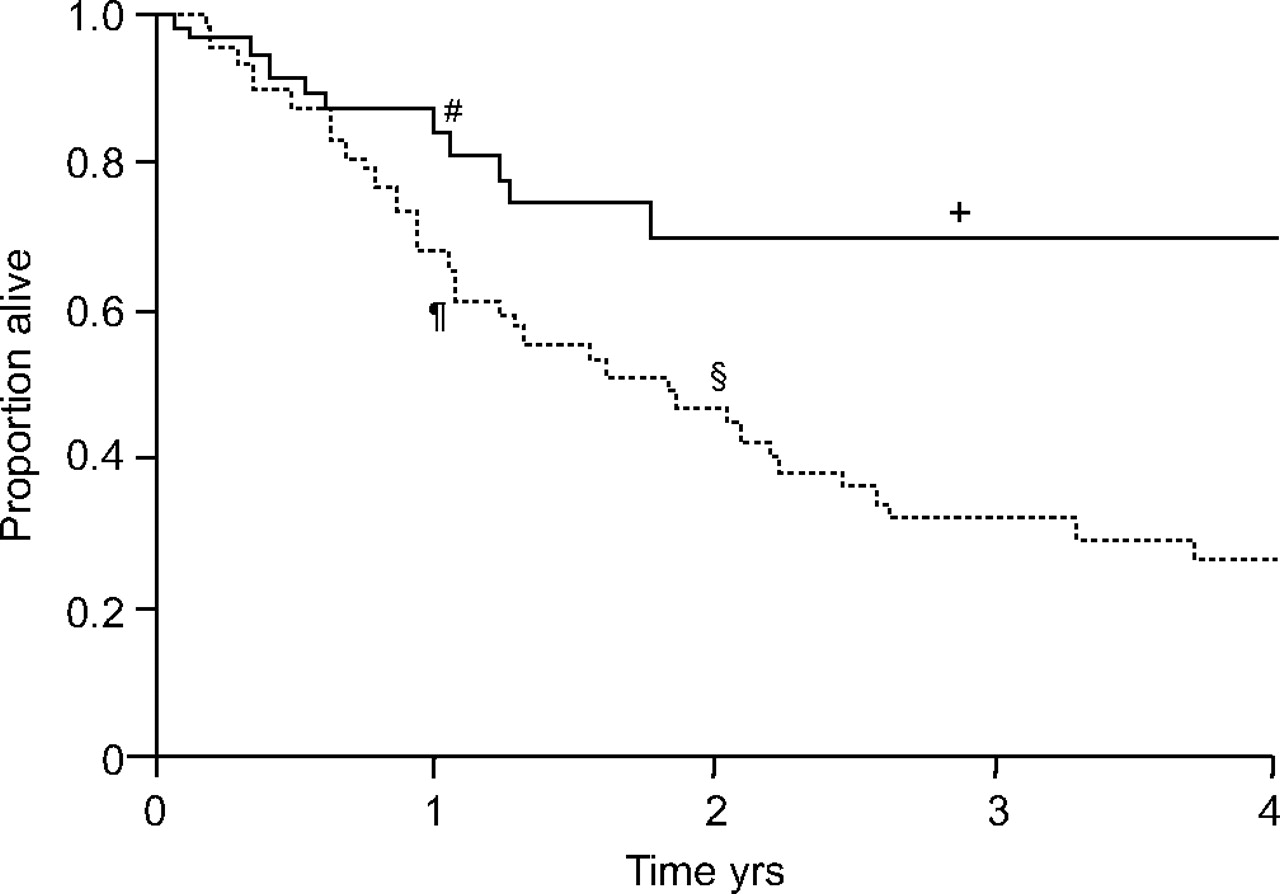
PAH is also an important complication of CTDs, such as systemic sclerosis (SSc). It is responsible for significant morbidity and mortality and represents a particularly aggressive form of PAH. Data from several clinical trials, as well as from usual clinical practice, demonstrate short- and long-term efficacy and tolerability of bosentan in this patient population [10–13]. In a long-term, open-label follow-up of the subgroup of PAH-CTD patients from the study by Channick et al. [4] and BREATHE-1, Kaplan–Meier estimates for observed long-term outcome compared favourably with published reports [14, 15]. These positive effects of bosentan on long-term outcome in patients with PAH associated with SSc (PAH-SSc) have been supported by the results from a 6-yr observational, longitudinal study, which compared long-term outcome in a cohort of PAH-SSc patients receiving bosentan as first-line therapy with a matched historical control group treated with conventional therapy, including prostanoids if needed (fig. 6⇓) [16].

Long-term outcome in 92 patients with pulmonary arterial hypertension-systemic sclerosis (PAH-SSc). Kaplan–Meier analysis showing mortality rates in patients with PAH-SSc. In the historical group (control ------; prior to the availability of bosentan) 1- and 2-yr survival rates were 68 and 47%, respectively. After the availability of bosentan (––––––) 1- and 2-yr survival estimates were 81 and 71%, respectively. #: 81%; ¶: 68%; +: 71%; §: 47%. Reproduced and modified from [16] with permission from the publisher.
The effects of bosentan on the quality of life (QoL) and severity of PAH in patients with PAH-CTD have also been investigated in a 48-week open-label study (TracleeR Use in PAH associated with Scleroderma and connective Tissue disease; TRUST). The results confirm the clinical benefits of bosentan in PAH-CTD patients. At the beginning of the study, all included patients were in NYHA FC III. After 48 weeks of bosentan therapy, a substantial proportion of patients improved their functional class, 57% remained stable and 16% worsened (fig. 7⇓) [17]. Stabilisation cannot be underestimated in a relentlessly progressive disease. Interestingly, QoL assessed using the nonspecific 36-item Short-Form Health Survey questionnaire improved significantly at week 16 and improvements were maintained at week 48 (table 3⇓). Kaplan–Meier estimates for survival were 92% (95% confidence interval 85–100%), which compares very well with available historical controls.

Percentage change in New York Heart Association functional class (NYHA FC) in pulmonary arterial hypertension (PAH) associated with connective tissue disease in TracleeR Use in PAH associated with Scleroderma and connective Tissue disease (TRUST) study. Of the 53 NYHA FC II patients enrolled in the TRUST study and treated with bosentan, 27% improved (▒), 57% remained stable (▓) and 16% deteriorated (░) over the course of the 48-week study.
36-item Short-Form Health Survey questionnaire health transition item: mean change from baseline
Congenital heart disease
Atrial septal defects, ventricular septal defects and complex heart defects can result in PAH. If these defects remain unrepaired, blood is shunted from the left to the right of the heart resulting in increased blood flow through the pulmonary arteries. With time, the lumen of pulmonary arterioles becomes narrower, due in part to shear stress-induced proliferation of multiple components of the vessel wall. These changes to the vasculature include endothelial and smooth muscle cell dysfunction and result in persistent elevation of PVR.
As the pressure in the pulmonary arteries increases, the shunt starts to reverse (right to left) with deoxygenated blood being forced back into the systemic circulation. This mixing of venous and arterial blood in the systemic circulation results in a reduction in overall oxygen saturation, which manifests clinically as cyanosis of the lips and fingertips.
The phenomenon of right to left shunting, together with persistently elevated PVR, is known as Eisenmenger's syndrome (ES) and represents a particularly severe form of PAH that is associated with extremely poor exercise tolerance and QoL, although prognosis is generally better compared with other forms of PAH. However, because the heart defect in ES is present from birth, the heart might be “primed” to deal with the right heart overload and is able to operate at systemic pressures. Studies in this group of patients have reported PVR measurements of 2,870–3,425 dyn·s·cm−5 compared with 880–1,167 dyn·s·cm−5 observed in patients with iPAH or PAH-CTD.
The histopathological features of ES are grossly identical to those observed in other forms of PAH, supporting the presence of a similar underlying pathophysiology. For instance, elevated levels of ET-1 have been measured in PAH-CHD [18], as well as in iPAH [19]. These findings formed the rationale behind the BREATHE-5 trial, which was the first randomised controlled trial in patients with ES and followed a series of encouraging open-label studies [20, 21].
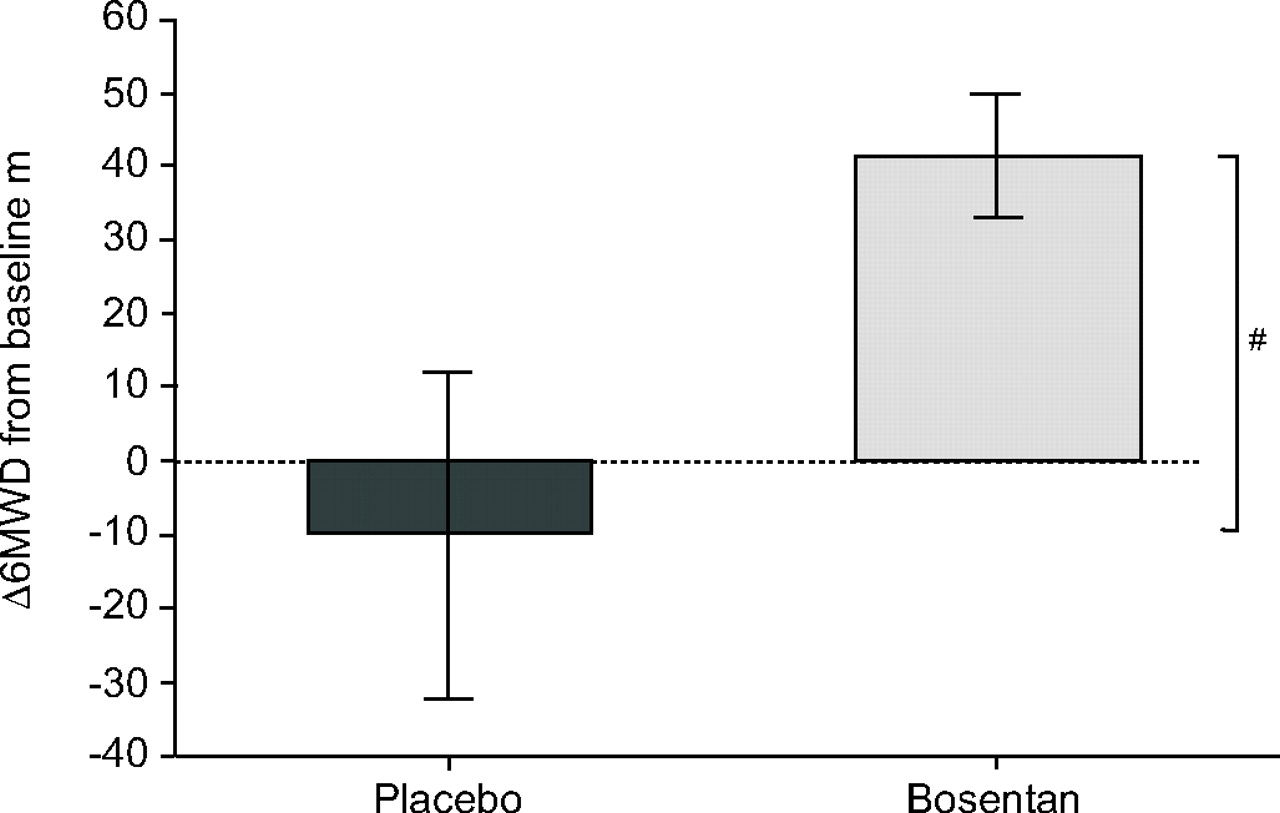
BREATHE-5 examined the effect of bosentan in ES patients after 16 weeks of double-blind therapy. The first objective was to demonstrate that bosentan did not aggravate the shunt and thus did not result in reduced oxygen saturation. This was found to be the case. In fact, a modest nonsignificant increase in oxygen saturation was evident and bosentan resulted in a significant reduction in PVR, an increase in exercise capacity (fig. 8⇓) and improved NYHA FC [22]. In an open-label extension to the BREATHE-5 study, improved exercise capacity was maintained in patients who continued to receive bosentan. In patients who received placebo in the double-blind phase of the trial, 6MWD improved by 33 m over the course of the open-label period when they were given bosentan [23].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Bosentan increased exercise capacity in patients with pulmonary arterial hypertension associated with congenital heart disease. Change from baseline in 6-min walk distance (Δ6MWD) in placebo (n = 17) and bosentan (n = 37) groups. Treatment effect = 53.1 m. #: p = 0.008. Reproduced and modified from [22] with permission from the publisher.
SAFETY OF BOSENTAN
Like all orphan drugs, the approval of bosentan was based on relatively few patient-yrs of therapy. Consequently, it was critical that comprehensive evaluation of safety was continued in post-marketing surveillance studies to obtain safety data from real-life usage. Tracleer PMS, a novel pharmacovigilance system, was set up in 18 countries to achieve just this and data are available from close to 5,000 patients, corresponding to ∼80% of all patients treated with bosentan in Europe [24]. Within 30 months, an elevation of liver aminotransferases more than three times the upper limit of normal values was observed in 7.6%, corresponding to an annualised rate of 10.1%. Safety results were consistent across subgroups and aetiologies. Importantly, no unexpected safety issues were detected and the well-characterised class effect of elevated liver aminotransferases was consistent with the levels reported in the pivotal randomised controlled trials [4, 5] and could be effectively managed using the system. Bosentan has now been administered to >30,000 patients worldwide.
ONGOING STUDIES
PAH is clearly a condition with a complex, multifactorial pathophysiology, which involves more than one signalling pathway [25, 26]. Future treatment strategies are likely to involve the use of more than one therapeutic agent, and investigations into the effects of combined treatment regimens are therefore essential. As a well-established treatment for PAH, it is envisaged that bosentan will be an important part of such regimens, as supported by preliminary data from the BREATHE-2 study [27]. Currently, two multicentre trials, COMPASS (Effects of Combination Of bosentan and sildenafil versus sildenafil Monotherapy on morbidity and mortality in symptomatic PAtientS with PAH)-1 and COMPASS-2, are investigating the combination of bosentan with the phosphodiesterase-5 inhibitor sildenafil and the possible additive effects of these two agents. COMPASS-1 is an open-label, assessor-blinded trial and COMPASS-2 is a double-blind, randomised, placebo-controlled study. COMPASS-2 is the only event-driven morbidity/mortality study in PAH. The COMPASS programme will contribute to present understanding of the optimal long-term management of PAH.
The effects of bosentan on exercise capacity and disease progression in patients with mildly symptomatic NYHA FC II PAH is also being investigated as part of the double-blind, placebo-controlled EARLY (Endothelin Antagonist tRial in mildLY symptomatic PAH patients) study, the only randomised, controlled trial designed to target mildly symptomatic NYHA FC II PAH patients, specifically. Further trials include BENEFiT (Bosentan Effects in iNopErable Forms of chronIc Thromboembolic pulmonary hypertension), a double-blind, placebo-controlled trial to evaluate the safety and efficacy of bosentan treatment in patients with inoperable chronic thromboembolic pulmonary hypertension and the ASSET (Bosentan to Treat Pulmonary Hypertension in Sickle Cell Disease) programme (ASSET-1 and ASSET-2) for the use of bosentan in pulmonary hypertension associated with sickle cell disease.
CONCLUSIONS
Extensive data from randomised clinical trials and their extensions, cohort studies and clinical practice have provided a wealth of clinical evidence showing that the dual endothelin receptor antagonist bosentan is an effective treatment modality for pulmonary arterial hypertension related to a number of conditions. In addition to providing symptomatic relief, such studies have shown bosentan to improve long-term outcomes in patients with pulmonary arterial hypertension. Research is ongoing to determine the effects of combining bosentan with other therapeutic agents and to investigate potential new applications of this therapeutic agent.
Statement of interest
M. Humbert has received: reimbursement for attending a symposium; fees for speaking; fees for organising education; funds for research; funds as a member of staff; and fees for consulting. M. Humbert has relationships with drug companies, including Actelion, Altana, Encysive, GlaxoSmithKline, Novartis, Schering, Pfizer and United Therapeutics. In addition to being an investigator in trials involving these companies, relationships include consultancy services and membership of scientific advisory boards.
- © ERSJ Ltd