





Abstract
A 41-yr-old patient with chronic stable idiopathic pulmonary arterial hypertension (PAH) presented with sudden chest pain and unusual dyspnoea during physical exertion. The patient had been diagnosed with PAH at the age of 12 yrs and was in New York Heart Association functional class I/II. The patient was being treated with an anticoagulant regimen, low-dose diuretics and continuous intravenous epoprostenol therapy.
A computed tomography scan showed ancient massive thrombi in dilated central pulmonary arteries, which were not haemodynamically significant (perfusion lung scans did not demonstrate segmental or larger defects), and extensive dissection of the right pulmonary artery starting from the intermediate branch. Due to the extensiveness of the dissection, the patient was immediately considered for heart–lung transplantation, but died 72 h after the onset of symptoms. Permission for post mortem examination was denied.
Pulmonary artery dissection should be suspected in PAH patients presenting with chest pain and worsening dyspnoea. In the current case, the factors possibly associated with increased risk for dissection may include dilatation of the pulmonary artery, local inflammation favoured by in situ thrombosis, and acute increase of pulmonary pressure secondary to physical exertion. Extensive pulmonary artery dissection is a life-threatening complication of PAH, and urgent heart/lung transplantation might be the treatment of choice in eligible patients. In addition, better identification of the risk factors for pulmonary artery dissection may help in considering transplantation for selected patients at risk.
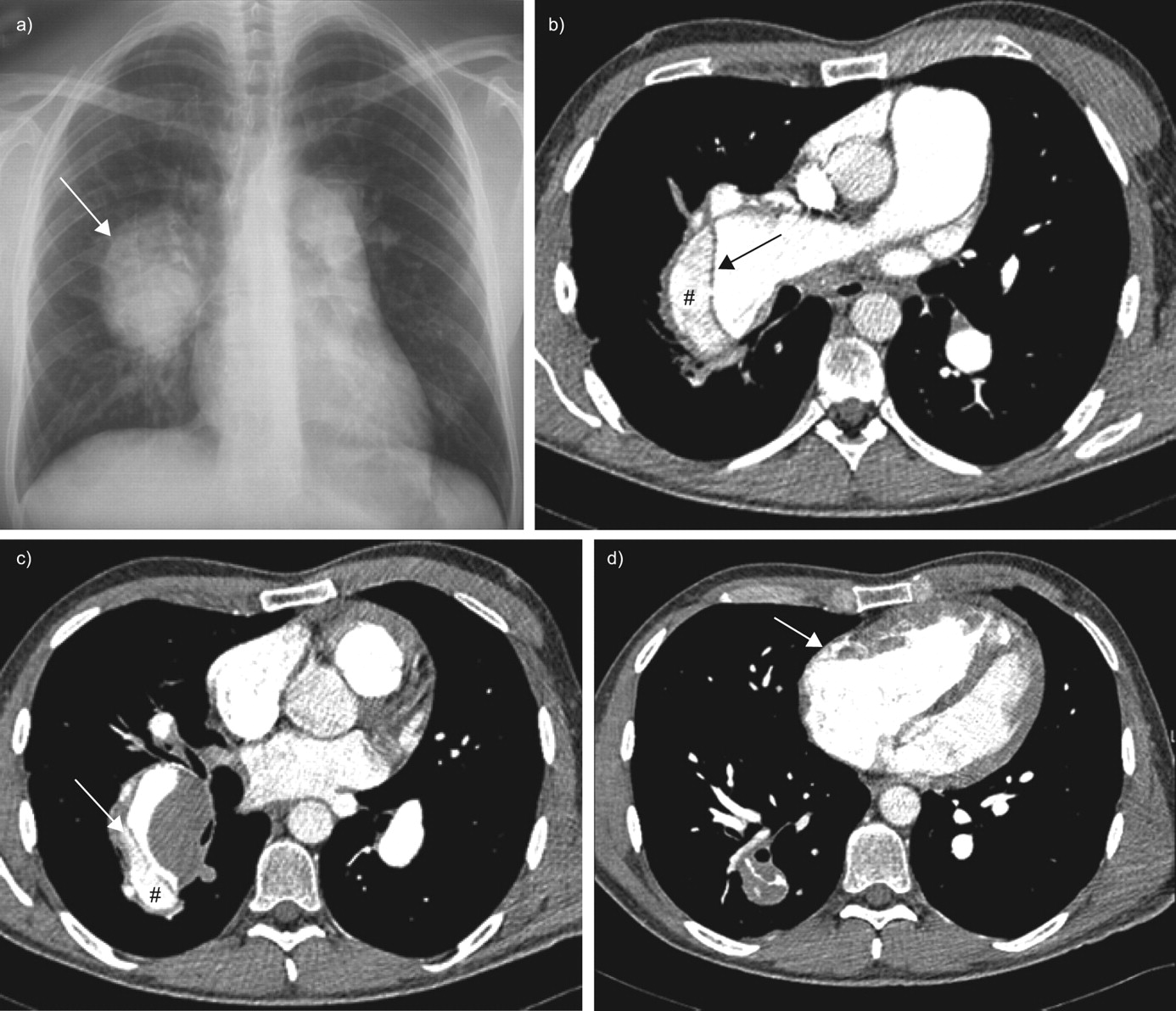
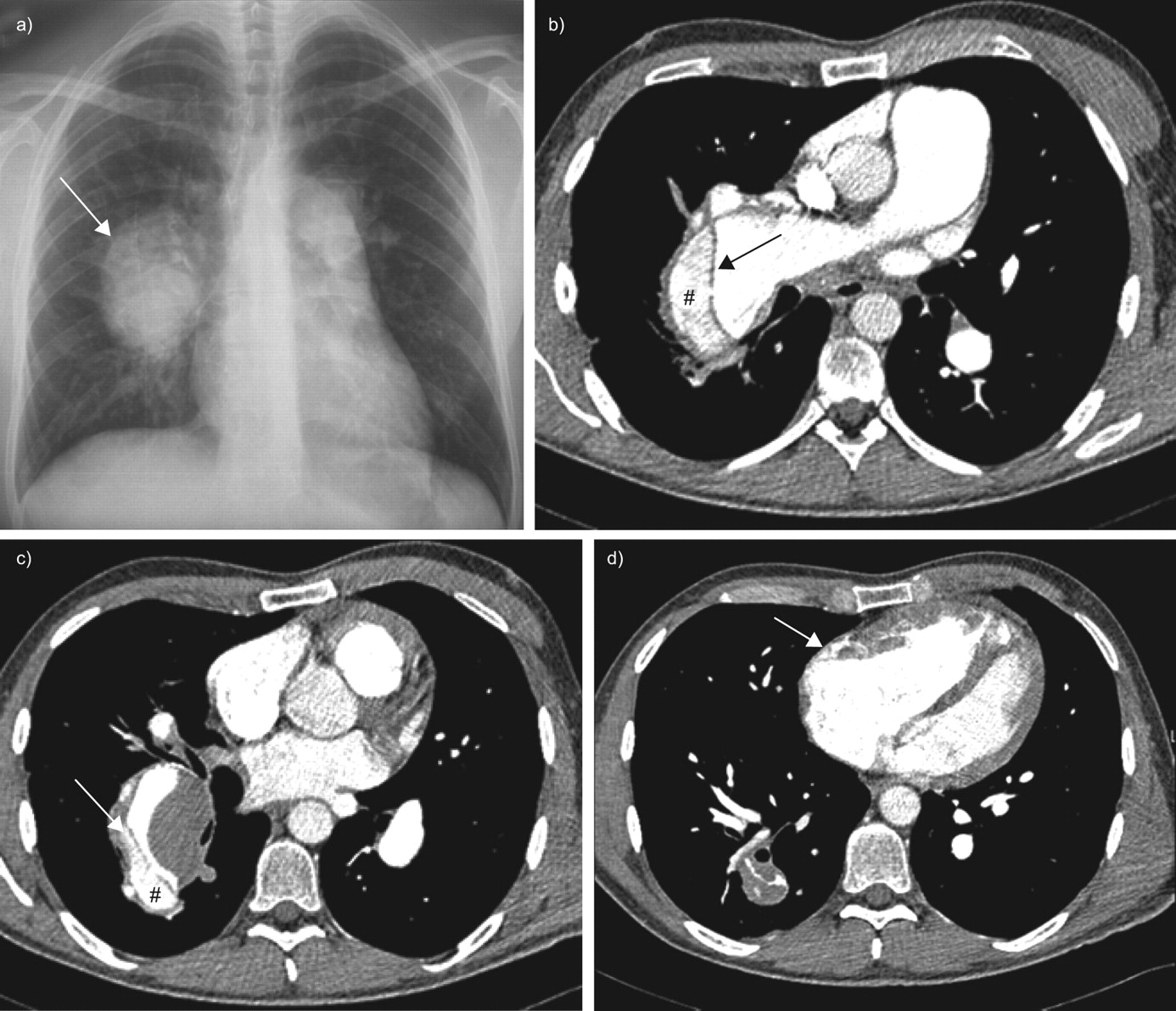
A 41-yr-old patient with idiopathic pulmonary arterial hypertension (PAH) was referred to the emergency department of the Rangueil-Larrey University Hospital (Toulouse, France) with sudden chest pain and worsening dyspnoea. This patient had idiopathic PAH without evidence of associated conditions or causes, such as congenital heart disease or chronic thromboembolic pulmonary hypertension, and had been diagnosed at the age of 12 yrs. He remained untreated and performed regular physical exercise without significant dyspnoea for the following 15 yrs. At the age of 27 yrs, he began to complain of exertional dyspnoea (New York Heart Association (NYHA) functional class II), but still remained untreated. At 30 yrs of age, he presented with increasing dyspnoea (NYHA functional class III) in association with physical signs of cardiac failure (pedal oedema and tachycardia). An oral anticoagulant and diuretics were prescribed. In addition, long-term treatment with intravenous epoprostenol was started; the patient received a continuous dose of 16–18 ng·kg−1·min−1 of epoprostenol for 8 yrs. During this period, he remained in NYHA functional class I/II and continued to perform regular physical exercise. In December 2004, when the patient was 38 yrs of age, treatment was switched from epoprostenol to i.v. treprostinil as part of a therapeutic trial. Approximately 1 yr after the change to treprostinil, the patient developed an upper left arm oedema. A chest radiograph showed hilar enlargement and dilatation of the right pulmonary trunk (fig. 1a⇓). A computed tomodensitometry (CT) scan of the chest revealed a catheter-related subclavian left vein stenosis; the catheter was free of any thrombosis. Therefore, the catheter was removed and placed in the right internal jugular vein. In addition, the CT scan of the chest revealed thrombosis of the intermediate branch of the right pulmonary artery (fig. 1b⇓). The thrombosis had developed despite the administration of anticoagulant treatment to maintain an international normalised ratio of 2.0–3.0. The large central thrombi were not haemodynamically significant and the perfusion lung scan did not demonstrate segmental or larger defects. The pulmonary artery thrombosis was considered to be a complication of the large dilatation of the artery, as previously described [1]. With this treatment, the patient remained in NYHA functional class I/II, but haemodynamic parameters deteriorated (cardiac index decreased from 3.5 to 2.5 L·min−1·m−2 and pulmonary vascular resistance increased from 730 to 1,020 dyn·s·cm−5), although the dose of treprostinil was increased to 65 ng·kg−1·min−1. Therefore, treatment was switched from treprostinil back to epoprostenol, and the dual endothelin-receptor antagonist bosentan was added to the treatment regime. The patient’s clinical status remained stable on this new treatment regimen, and haemodynamic parameters did not improve. Less than a year later, the patient developed acute chest pain and severe dyspnoea when he was cycling. He was referred to the emergency department <4 h after the onset of symptoms. On clinical examination, he had a blood pressure of 95/65 mmHg, heart rate of 125 beats·min−1, respiratory rate of 31 breaths·min−1 and hepatojugular reflux. A full blood count, serum urea and electrolyte concentrations, and his C-reactive protein concentration, were all normal. Liver function tests showed alanine aminotransferase and aspartate aminotransferase of two-fold the upper limit of normal values. Arterial blood gases on room air displayed a pH of 7.48, arterial oxygen tension of 5.9 kPa, arterial carbon dioxide tension of 3.4 kPa and oxygen saturation of 78%. ECG showed a complete right branch block and right axis deviation. A chest radiograph showed a marked increase in the enlargement of the right pulmonary trunk (fig. 2a⇓). A CT scan of the chest demonstrated extensive dissection of the right pulmonary artery starting from the intermediate branch and progressing to subsegmentary branches of right middle and lower lobe artery (fig. 2b⇓ and c). The starting point of the dissection was in the area of the previously noted pulmonary artery thrombosis. There was no evidence of pericardial effusion (fig. 2d⇓). The patient received symptomatic treatment with analgesics and supplemental oxygen. Because of the extensiveness of the dissection, heart/lung transplantation was considered to be the only therapeutic option, but the patient died suddenly 72 h after the onset of symptoms. Permission for post mortem examination was not granted.

a) Chest radiograph performed 8 months before the acute pulmonary artery dissection showed dilatation of the right pulmonary trunk (arrow). b) At the same time, the computed tomodensitometry scan of the chest revealed a thrombosis of the intermediate branch of the right pulmonary artery (arrow).
{kind=link}
{kind=link}
a) At the time of acute chest pain and dyspnoea, the chest radiograph showed a marked increase of the enlargement of the right pulmonary trunk (arrow). b and c) Computed tomodensitometry scans of the chest demonstrating extensive dissection of the right pulmonary artery starting from the intermediate branch and progressing to subsegmentary branches of right middle and lower lobe artery. The arrows indicate the intimal flap. #: the false lumen within the pulmonary arterial tree. d) There was no evidence of pericardial effusion on the computed tomodensitometry scan. The arrow indicates the enlarged right ventricle.
DISCUSSION
Pulmonary artery dissection is a highly life-threatening complication of PAH. Indeed, the false lumen in pulmonary artery dissection tends to rupture rather than develop a re-entry site (as is usual in aortic dissection), resulting in pericardial tamponade or major bleeding and subsequent death [2]. Of note, a large majority of pulmonary artery dissections are diagnosed post mortem [3]. As a necropsy is not always performed in PAH patients who have died suddenly, the frequency of this complication as a cause of sudden death in chronic PAH might be underestimated.
Symptoms of pulmonary artery dissection are nonspecific, such as chest or interscapular pain, dyspnoea and cyanosis. Chest radiographs may show mediastinal enlargement, pleural effusion, hilar masses and/or parenchymal opacities. Echocardiography may be useful to visualise a proximal dissection and/or a complication, such as pericardial effusion. Pulmonary arteriography, CT scans and magnetic resonance imaging of the chest are key diagnostic tools that are able to detect an intimal flap and a false lumen within the pulmonary arterial tree, and are able to evaluate the extensiveness of the lesions [4–7]. It was suggested that a pseudo-flap in the pulmonary artery may represent motion artefact from aortic and cardiac pulsation motion, and thus could lead to false diagnosis of pulmonary artery dissection [8]. If such an artefact is suspected, confirmation using a CT scan with ECG gating should be considered.
In a pulmonary vessel, an intimal tear can develop either spontaneously or after a wound caused by a pulmonary artery catheter [9]. However, in a recent review of >7,000 procedures performed in expert pulmonary vascular centres in the modern management era, no case of pulmonary artery dissection was reported [10]. However, in this review, rupture of the pulmonary artery was reported in one case during an elective procedure with a balloon-tipped catheter [10]. Although the pathophysiological cause of spontaneous pulmonary artery dissection in patients with pulmonary hypertension is not clear, medial degeneration with fragmentation of elastic fibres of the large elastic pulmonary arteries and marked pulmonary arterial dilatation strongly predispose to this condition [2, 11, 12]. In PAH, dissection most often occurs at the site of a pulmonary aneurysm or a marked dilatation [2]. It is reasonable to assume that a spontaneous dissection occurs at the point where pulmonary artery tissue becomes too fragile to support the tension of the pulmonary artery wall. In dilated vessels of PAH patients, the increase of pulmonary pressure along with a large radius of the vessels led to increased parietal wall tension, as predicted by Laplace's law [13]. In addition to these conditions, pre-existing local inflammation of the pulmonary vessels caused by thrombosis may also be related to the formation of intimal tear [12]. Several of these potential causes coexisted in the case presented herein. The patient had very dilated pulmonary arteries, because of the long-lasting history, as previously demonstrated in patients with congenital heat disease-associated PAH [14]. He may also have experienced increased pulmonary artery pressure and subsequent increased parietal wall tension as a result of physical exercise, as he was cycling when his symptoms began. Moreover, he had a large pulmonary artery thrombosis in the area where the tear occurred, which could have contributed to weakening of the artery wall.
Therapeutic options in pulmonary artery dissection are scarce. Symptomatic treatments may include cough suppressants, analgesics, oxygen, vasodilators, diuretics and dobutamine [3, 15]. However, the only curative therapeutic option is surgery. Senbaklavaci et al. [2] reported a case of surgical repair of a dissection of the main pulmonary artery resulting from PAH. The CT scan showed that due to the extensiveness of the dissection surgery could be performed. The patient was transferred to the operating room with signs of imminent cardiac tamponade and shock. The time interval between the onset of symptoms and surgery was ∼24 h. A T-shaped prosthetic graft replacement of the central pulmonary artery was successfully performed using the inclusion technique under cardiopulmonary bypass. Because of persistent PAH, the patient underwent double lung transplantation 9 months after the emergency operation. Wuyts et al. [15] reported a case of extensive dissection of the pulmonary artery in a patient with PAH. Pleural effusion was present at the time of diagnosis of pulmonary dissection. Since being diagnosed with pulmonary arterial dissection, this patient had several episodes of heavy chest pain, accompanied with an increase of troponin levels and of serum lactate. When pleural effusion increased, dobutamine was administered and associated with the patient’s recovery. A heart/lung transplantation was successfully performed after the patient had been on the urgent waiting list for 7 weeks. Steurer et al. [7] reported a case of pulmonary artery dissection in a 64-yr-old female patient with severe PAH. A diagnosis of pulmonary artery dissection was suggested following echocardiography and was confirmed by pulmonary arteriography. Follow-up of this patient after 1 yr was uneventful. However, in other cases, as in our observation, patients died during the initial hours following the first clinical signs [16].
According to the very poor outcome of PAH patients who develop pulmonary artery dissection, identification of patients at risk for this complication may be of interest in clinical practice. Pulmonary artery dilatation and pulmonary aneurysm are the most frequently reported factors in pulmonary artery dissection [2, 3, 7, 11, 17, 18]. In PAH, pulmonary artery dilatation and aneurysm have been associated with other complications, such as hoarseness (secondary to left recurrent laryngeal nerve palsy) [19], pseudo-asthma (secondary to the compression of the proximal bronchi) [20] and angina or left ventricular ischaemia (due to extrinsic compression of the left main coronary artery) [21]. These complications seem directly related to the size of the pulmonary arteries. For example, pulmonary trunk diameter is correlated with the likelihood of left coronary artery compression; therefore, detection and appropriate treatment of this life threatening complication was proposed in selected patients based on noninvasive measurement of pulmonary artery diameter [22]. Dissection and rupture of a dilated pulmonary artery is a rare event which is most difficult to predict. Laplace's law describes parietal wall tension as being dependent on the pressure and diameter of the vessel. In PAH patients with dilated pulmonary vessels and preserved cardiac function, the increase of pulmonary pressure during an effort [23] leads to an increase in parietal wall tension that could lead to an excess risk of development of an intimal tear. Whether or not the presence of a parietal thrombosis may increase the risk of dissection remains to be determined. As in situ thrombosis of dilated central pulmonary arteries is a common feature of congenital heart disease-associated PAH and pulmonary artery dissection a rather rare event, one must consider that the role of local thrombosis in promoting dissection remains to be demonstrated [14]. Better identification of risk factors for pulmonary artery dissection may help to consider transplantation for selected patients at risk, as well as provide recommendation regarding physical exercise limitation.
Statement of interest
M. Humbert has relationships with drug companies including Actelion, Bayer Schering, GlaxoSmithKline, Novartis, Pfizer and United Therapeutics. In addition to being investigators in trials involving these companies, relationships include consultancy services and membership of scientific advisory boards.
Provenance
Submitted article, peer reviewed.
- Received May 24, 2009.
- Accepted May 29, 2009.
- © ERSJ Ltd
References









