





Abstract
A 63-yr-old black female, with a 1-yr history of hepatitis C and ascites was referred to an expert centre with suspicion of portopulmonary hypertension (PPHTN). Her poor condition made a rapid diagnosis imperative and precluded a normal diagnostic work-up.
Echocardiography confirmed severe pulmonary hypertension (PH). A hepatic scintigraphy and an abdominal echo-Doppler study excluded liver cirrhosis and portal hypertension. Cardiac magnetic resonance imaging showed marked dilation of the right ventricle with significant hypertrophy of the free wall, a finding that is uncommon in idiopathic pulmonary arterial hypertension or PPHTN. Right heart catheterisation demonstrated severe pre-capillary PH without response to acute vasodilator testing. Finally the patient underwent computed tomography angiography, which showed marked dilation of the pulmonary artery without thromboembolic disease and, unexpectedly, a partially calcified large patent ductus arteriosus.
The correct diagnosis of the underlying cause of pulmonary arterial hypertension is essential. Patients with underlying heart defects may have an atypical presentation and be referred to expert centres with an incorrect diagnosis. A full investigation is necessary; careful examination of right ventricular anatomy can provide clues about the aetiology of PH, and it is important to exclude intra- and extracardiac shunts during haemodynamic studies.
The patient was a 63-yr-old black female who had a history of eight pregnancies with three abortions and hepatitis C virus-related hepatic cirrhosis, which was diagnosed in 2006. During 2007, she was frequently hospitalised for ascites. She first presented in 2007 when investigation using transthoracic Doppler echocardiography demonstrated a very high estimated systolic pulmonary artery pressure of 130 mmHg. She was referred to our centre (Pulmonary Hypertension Unit, Dept of Cardiovascular and Respiratory Disease, Sapienza University of Rome, Rome, Italy) on suspicion of portopulmonary hypertension (PPHTN).
On admission to our centre, the patient was symptomatic at rest (World Health Organization functional class III/IV) and in very poor general condition, with hypotrophic muscular mass. There were signs of overt right heart failure, with jugular distension (4–5 cm), hepatomegaly and ascites, and she was unable to perform the 6-min walk test. Arterial blood gases showed hypoxaemia (56 mmHg) with hypocapnia (33 mmHg).
The poor condition of the patient made a rapid diagnosis imperative and precluded the usual diagnostic work-up. Earlier medical records were largely missing due to her immigrant status.
Echocardiography confirmed severe pulmonary hypertension (PH). There was significant right ventricular dilation (diastolic diameter 60 mm) with severe right ventricular free wall hypokinesis (tricuspid annular plane systolic excursion 12 mm) and paradoxical movement of the interventricular septum. Global systolic ventricular function was depressed (ejection fraction (EF) 40%) with severe tricuspid insufficiency (right ventricular–right atrial gradient 117 mmHg). There was also severe main pulmonary artery dilation with a suspected image of intravascular thrombus at the origin of the left pulmonary artery.
Hepatic cirrhosis was excluded by hepatic scintigraphy and abdominal Doppler echocardiography was used to exclude portal hypertension.
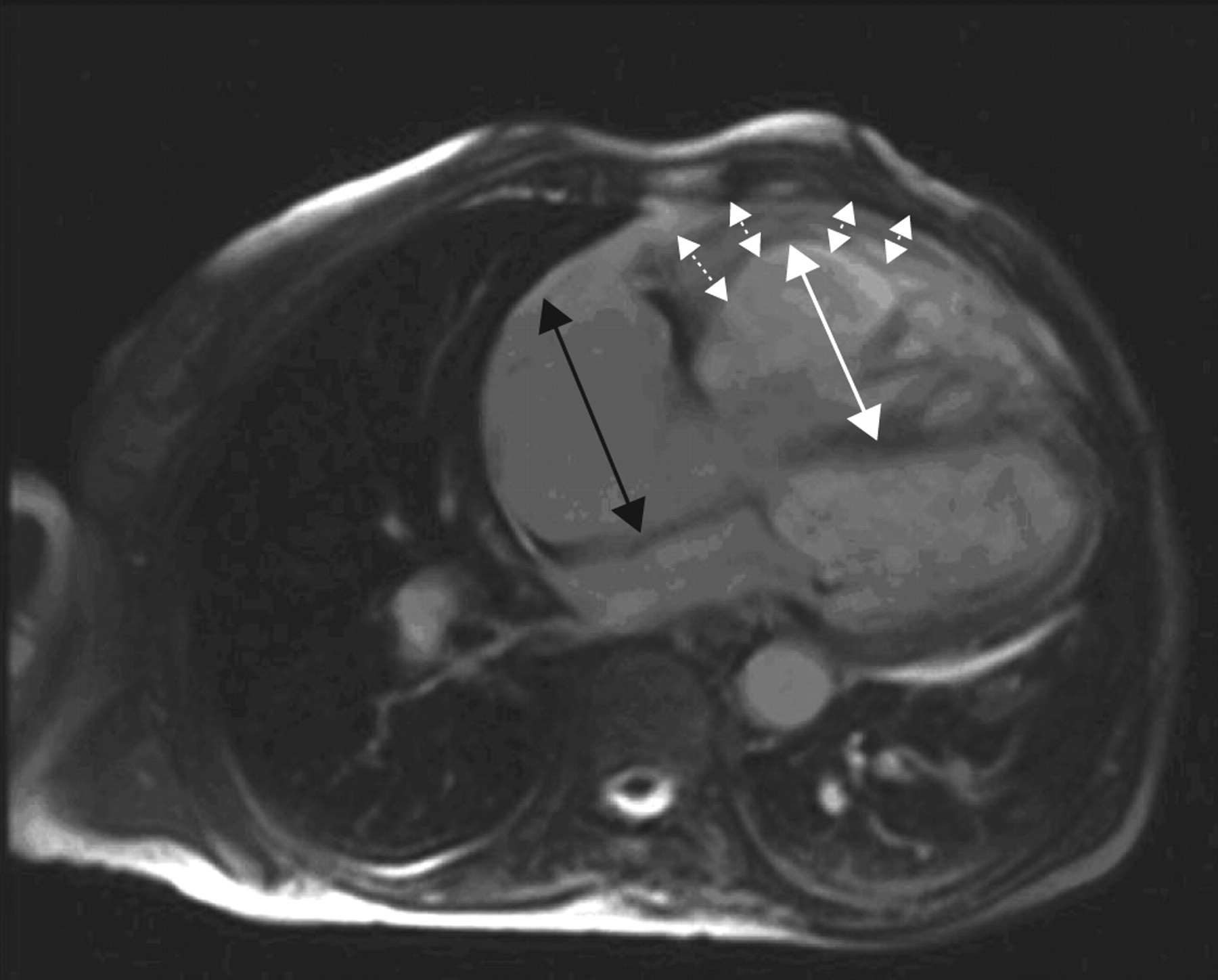
Magnetic resonance imaging (MRI) of the heart (fig. 1⇓) showed marked dilation of the right ventricle with significant hypertrophy of the free wall. There was severe systolic dysfunction and reduced global kinesis (EF 35%), no compression of the left ventricle by the right ventricle, with paradoxical movement of the interventricular septum. There was mild pericardial effusion but no thrombotic apposition in the pulmonary artery or in the main branches was seen. At this point, there was some suspicion of congenital heart disease (CHD; marked right ventrical free-wall hypertrophy). Doppler echocardiography excluded the presence of an intracardiac shunt; the quality of angio-MRI was insufficient to exclude extracardiac shunt.
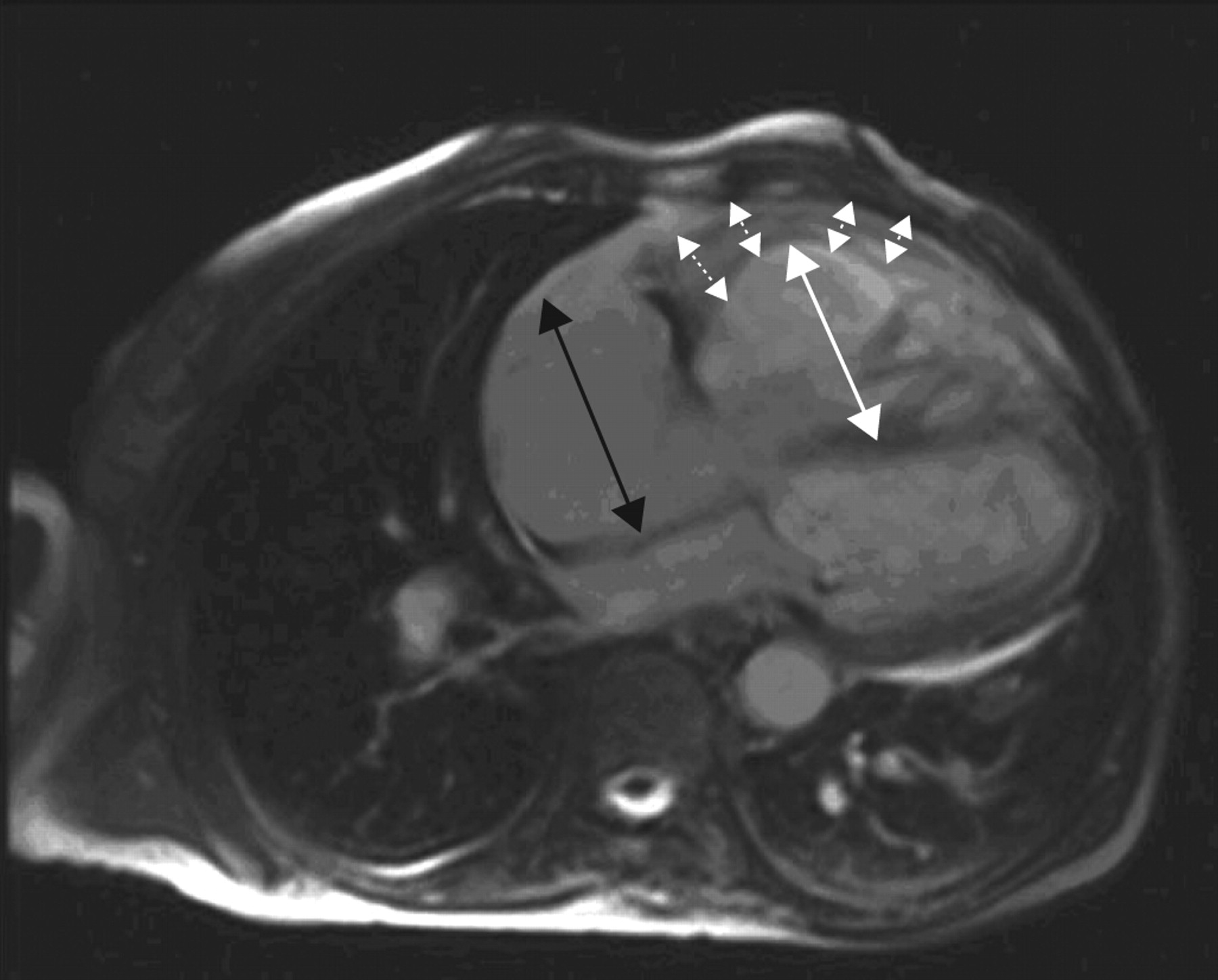
Cardiac magnetic resonance imaging showing significant hypertrophy of the right ventricle (dotted arrows) and severe dilation of right ventricle (white arrow) and atrium (black arrow).
Right heart catheterisation demonstrated severe pre-capillary PH (mean pulmonary artery pressure 83 mmHg, mean pulmonary wedge pressure 11 mmHg) with high right atrial pressure (mean right atrial pressure 17 mmHg) and a very low cardiac index (1.5 L·min−1·m−2). There was no response to nitric oxide. At this time several elements were discordant: 1) the patient did not have portal hypertension; 2) there was marked right ventricular free-wall hypertrophy (less frequent in idiopathic PH or PPHTN); and 3) there was marked pulmonary artery dilation (a finding that could be present in idiopathic or PPHTN [1], but that is more common in Eisenmenger syndrome).
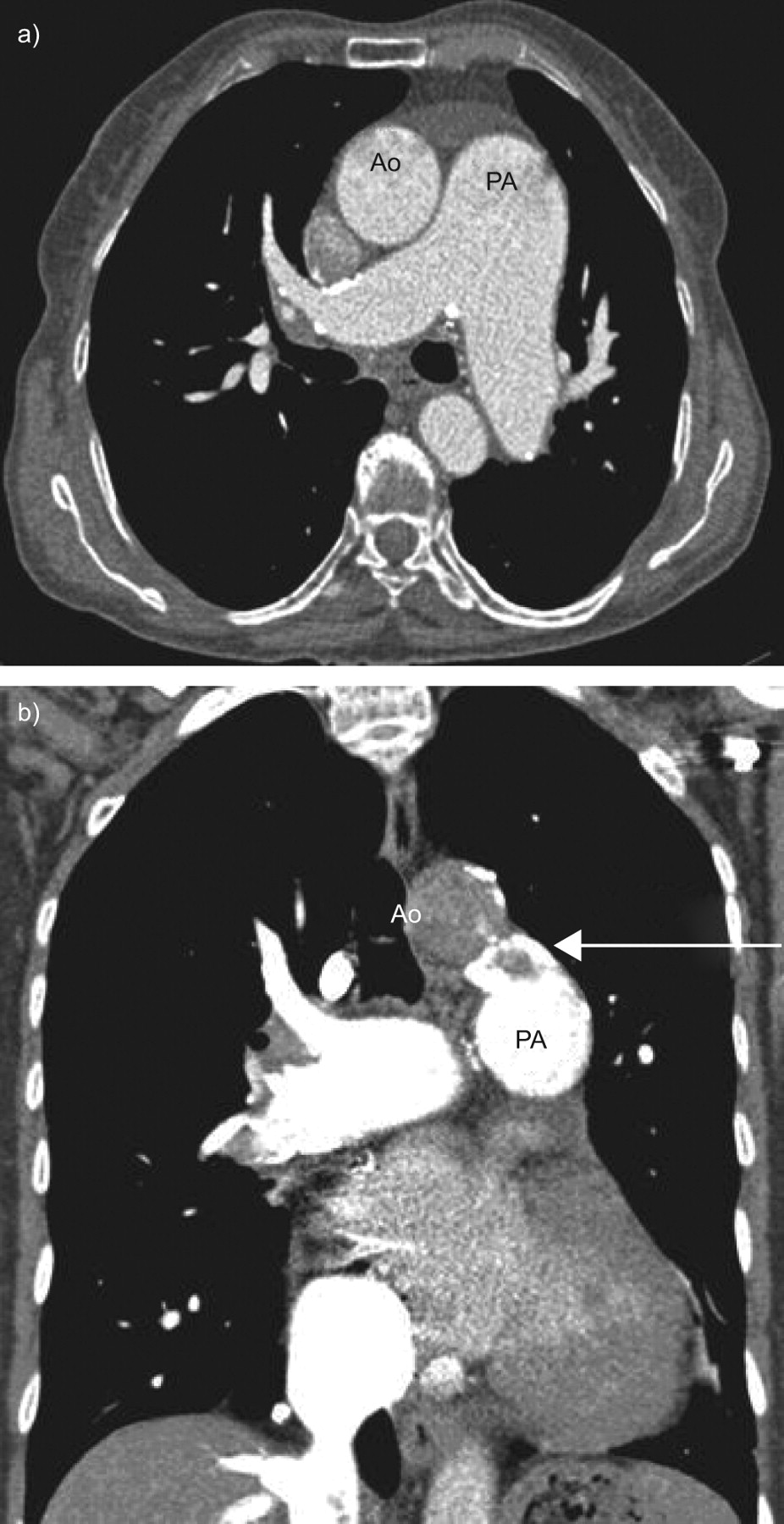
Computed tomography angiography (fig. 2⇓) was conducted in order to exclude thromboembolic disease; a ventilation/perfusion scan was not immediately available. This showed marked dilation of the pulmonary artery (pulmonary artery/aorta ratio 1.8), but also a partially calcified large patent ductus arteriosus (∼15 mm diameter) between the aortic isthmus and the origin of the left pulmonary artery. Therefore, this patent ductus arteriosus was the cause of PH.

{kind=link}
{kind=link}
a) High-resolution computed tomography showing marked pulmonary artery dilatation, and b) angio-computed tomography showing a patent ductus arteriosus (arrow). Ao: aortic arch; PA: pulmonary artery.
Considering her clinical condition and the haemodynamic assessment, treatment with subcutaneaous treprostinil was initiated, the dose increased without issue, and the patient was discharged in a much better clinical condition: reduction in ascitis and improvement in effort capacity with a distance at the 6-min walk test of 240 m. She remained in a clinical stable condition for a further 6–8 months, but then died suddenly.
DISCUSSION
This case highlights the potential for misdiagnosis of the underlying cause of PH in adult patients, especially among those with CHD. Always progressive and frequently fatal, PH has a range of aetiologies [2]; diagnosis and classification of the underlying cause is vital, as different treatment options are available for different aetiologies and stages of PH. For pulmonary arterial hypertension (PAH), endothelin receptor antagonists, phosphodiesterase type-5 inhibitors and prostacylin analogues are the mainstays of treatment [3], although bosentan is the only therapy to have been evaluated in a randomised controlled trial dedicated to patients with PAH-CHD and Eisenmenger's syndrome [4].
Dyspnoea is the most common presenting symptom of PAH, regardless of aetiology, but there is a wide range of possible manifestations which, along with some atypical findings, can complicate diagnosis. Typical clinical signs of PAH-CHD include central cyanosis, clubbing, peripheral oedema, abdominal tenderness, right ventricular heave, a loud pulmonary ejection click and an accentuated pulmonary component of the second heart sound [5]. However, patients with PAH-CHD may present atypically. In this case, the atypical clinical presentation of patent ductus arteriosus was complicated by the absence of detailed records from a young age, the late onset of symptoms at 60 yrs of age, and the fact that cyanosis was not obvious due to the patient's skin colour.
As described here, patients with PAH may be referred to expert centres with an incorrect diagnosis. In a patient with liver disease, ascites very often leads to the suspicion of cirrhosis when this may instead be due to severe right ventricular dysfunction. This makes a full investigation, conducted on admittance, a necessity. Careful examination of right ventricular anatomy can provide clues about PAH aetiology; in this case there was significant hypertrophy of the free-wall and the left ventricle was not compressed by the large right ventricle. Small intraventricular defects may not be evident on echocardiography, so intra- and extracardiac shunts should always be excluded by haemodynamic studies. In the present case, oximetry along the right ventricle and the pulmonary artery was not performed.
Conclusions
PAH is an aggressive disease and early diagnosis and treatment is of the utmost importance, particularly in those forms for which treatment is available. The correct diagnosis of the underlying cause of PAH is essential but is complicated by the wide range of clinical manifestations associated with the disease and their potential for misinterpretation. Patients with PAH may be referred to expert centres with an incorrect diagnosis, and so full investigation should be performed. Misdiagnosis of patients with PAH can lead to substantial morbidity and may be potentially life-threatening, and the potential for misdiagnosis and for atypical presentation should always be considered.
Statement of interest
C.D. Vizza has received lecture fees paid by a commercial sponsor: Actelion, \#8364;5,000 in 2007–2008; Pfizer, \#8364;1,500 in 2008; and GlaxoSmithKline, \#8364;3,000 in 2007–2008. C.D. Vizza served on the advisory board of United Therapeutics in 2008 (\#8364;2,000) and GlaxoSmithKline in 2007–2009 (\#8364;3,500).
Provenance
Publication of this peer‐reviewed article was supported by Actelion Pharmaceuticals Ltd, Switzerland (unrestricted grant, European Respiratory Review issue 113).
Acknowledgments
We would like to thank A. Gray from Elements Communications Ltd (Westerham, UK) for medical writing assistance supported by Actelion Pharmaceuticals Ltd (Allschwil, Switzerland).
- Received June 2, 2009.
- Accepted June 8, 2009.
- © ERSJ Ltd









