





Abstract
A 29-yr-old patient with hereditary haemorrhagic telangiectasia was referred to the present authors’ centre with progressive exertional dyspnoea.
Pulmonary arterial hypertension (PAH) was suspected on Doppler echocardiography and confirmed by right heart catheterisation demonstrating severe PAH. Genetic analysis found an activin receptor-like kinase-1 gene missense mutation. Chest radiography and computed tomodensitometry of the chest revealed a pulmonary arteriovenous malformation with a 5-mm diameter feeding artery in the right lower lobe. Embolisation of the arteriovenous malformation was discussed, but was considered a very high-risk procedure that could aggravate PAH and was therefore not performed. Haemodynamics were improved by dual endothelin receptor antagonist and inhaled iloprost but the patient subsequently died suddenly of a rupture of the arteriovenous malformation into the pleural cavity.
Severe PAH is generally considered a contraindication to performing pulmonary arteriovenous malformation embolisation because of the risk of worsening of PAH. However, given the significant risk of rupture, paradoxical embolism and haemoptysis, and the lack of data regarding the evolution of pulmonary pressure after embolisation in PAH, pulmonary arteriovenous malformation embolisation should not be absolutely contraindicated and might be considered in patients with stable PAH.
- Activin receptor-like kinase-1
- arteriovenous malformation
- embolisation
- hereditary haemorrhagic telangiectasia
- pulmonary arterial hypertension
- rupture
A 29-yr-old patient with hereditary haemorrhagic telangiectasis (HHT) was referred to the present authors’ centre (Hôpital Antoine-Béclère, Assistance Publique – Hôpitaux de Paris, Clamart, France) with suspected pulmonary arterial hypertension (PAH). HHT was diagnosed at age 10 yrs in the context of familial HHT (fig. 1⇓), repeated epistaxis and telangiectasia involving skin and mucous membranes. In 2001, the patient became pregnant, and although she had no obstetric complications during the pregnancy, shortly after delivery she presented with a transient right hemiplegia. In January 2003, she developed rapidly progressive exertional dyspnoea (New York Heart Association (NYHA) functional class II) with two episodes of dizziness. Her dyspnoea increased progressively and, 6 months later, she was described as NYHA functional class III. On clinical examination there was a prominent pulmonary component of S2 with a systolic murmur suggesting tricuspid regurgitation. ECG showed a complete right branch block and right axis deviation. Spirometry was normal except for a mild decrease in diffusion capacity of the lung for carbon monoxide. Arterial blood gases showed oxygen tension (PO2) of 80 mmHg (11 kPa) and carbon dioxide tension 32 mmHg (4.3 kPa) in the supine position with hypoxaemia (PO2 72 mmHg (9.6 kPa)) on standing. Chest radiograph (fig. 2⇓) and computed tomodensitometry (fig. 3⇓) of the chest revealed a pulmonary arteriovenous malformation (PAVM) with a 5-mm diameter feeding artery in the right lower lobe. Transthoracic contrast echocardiography showed an estimated systolic pulmonary artery pressure (Ppa) of 70 mmHg, paradoxical septal motion and an extracardiac right-to-left shunt (with the appearance of microbubbles in the left atrium after four cardiac cycles). The presence of a PAVM could have explained the episode of transient right hemiplegia after delivery due to a paradoxical embolism. Computed tomography of the abdomen suggested a small hepatic arteriovenous malformation. Ventilation and perfusion lung scan did not show perfusion defects. The 6-min walk distance (6MWD) was 412 m with a Borg dyspnoea index of 2.5 and a reduction in arterial oxygen saturations from 94% to 87% on air. Right heart catheterisation confirmed severe PAH with mean Ppa of 50 mmHg and decreased cardiac index (table 1⇓). Genetic analysis found a missense mutation of activin receptor-like kinase-1 (ALK-1) gene (exon 9, c.1280A>T, p.Asp427Val). Embolisation of the arteriovenous malformation was discussed, but was considered a very high-risk procedure that could aggravate the patient’s pulmonary hypertension and was therefore not performed. Specific PAH therapy with the dual endothelin receptor antagonist bosentan was initiated. She was not anticoagulated because of repeated epistaxis. When evaluated after 4 months of therapy, NYHA functional class and 6MWD had improved (table 1⇓). Haemodynamics showed a decreased mean Ppa (-24%) and pulmonary vascular resistance index (-43%) with normal cardiac index (table 1⇓). After 12 months, clinical and haemodynamic parameters were stable but, after 18 months, the patient had an increase in exertional dyspnoea in functional class III associated with haemodynamic worsening (table 1⇓). Inhaled iloprost was therefore added to bosentan therapy, with an improvement after 3 months. However, the patient presented at emergency room with respiratory distress, collapse, right-sided chest pain and massive right-sided haemothorax. The patient died a few hours later, despite intensive care management. The presumed cause of death was a rupture of the arteriovenous malformation into the pleural cavity.

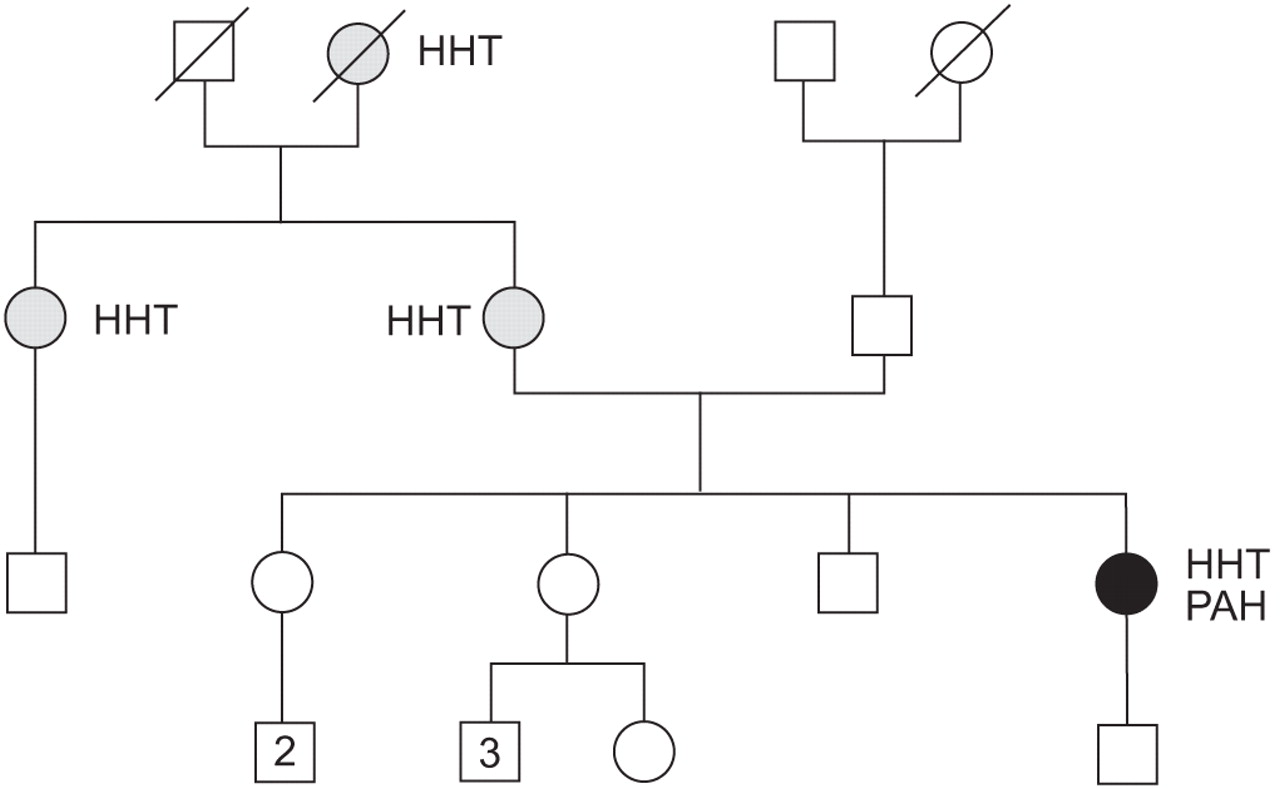
Familial history of hereditary haemorrhagic telangiectasis (HHT) of the 29-yr-old patient (•) with HHT and severe pulmonary arterial hypertension (PAH). Familial history analysis showed three other cases of HHT (▓) and no case of PAH. Genetic analysis of the patient found a missense mutation of the activin receptor-like kinase-1 gene.

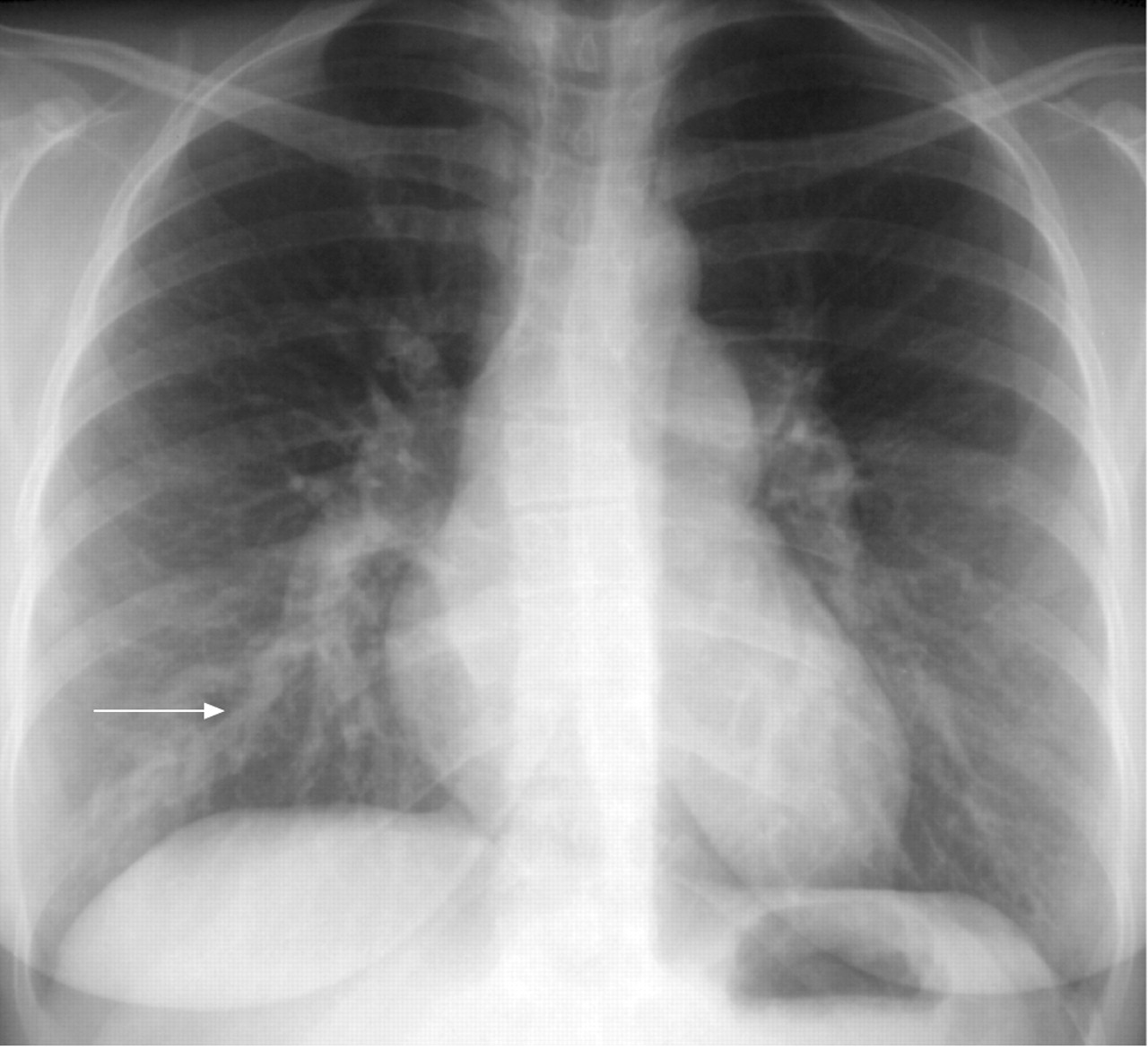
Chest radiograph revealed enlargement of the two pulmonary arteries and the presence of an opacity in the right lower lobe (arrow).

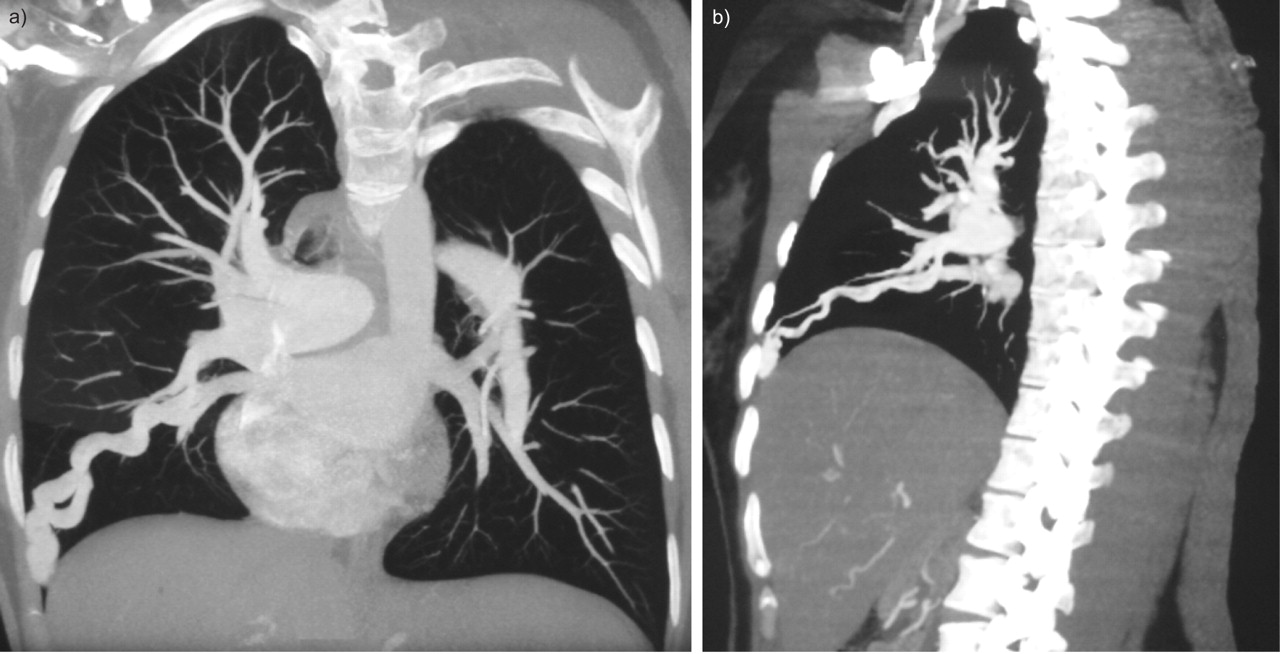
Computed tomodensitometry of the chest revealed a pulmonary arteriovenous malformation with a 5-mm diameter feeding artery in the right lower lobe.
Evolution of functional and haemodynamic characteristics
DISCUSSION
PAVMs are infrequent in the general population and most PAVMs occur in the context of HHT [1–4]. HHT is an orphan disorder of genetic origin, characterised by epistaxis, mucocutaneous telangiectasia and vascular visceral involvement, with arteriovenous malformations in various organs, especially the lung. PAVMs consist of an abnormal communication between pulmonary arteries and pulmonary veins, leading to right-to-left shunt, and are usually present in the lower lobes of the lungs [1, 2, 5]. Their prevalence in patients with HHT is estimated to be 15–33% [1, 2] and transthoracic contrast echocardiography may be useful for screening for PAVMs in HHT [6]. The main complications in HHT are due to PAVMs, with chronic hypoxaemia related to the right-to-left shunt, paradoxical embolic strokes, brain abscesses, haemoptysis and rupture (leading to massive haemoptysis and/or haemothorax). Embolisation of PAVMs is considered a safe procedure in HHT and an effective method of improving oxygenation and exercise tolerance, and reducing risk of paradoxical emboli and cerebral complications, and theoretically reduces the risk of rupture [2, 4, 5].
HHT is inherited as an autosomal dominant transmission with late-onset penetrance [7]. Several genes have been implicated in the pathogenesis of HHT. These include ALK-1, located on chromosome 12, endoglin on chromosome 9, MADH4 (encoding SMAD4, whereby mutations also lead to juvenile polyposis) [8] and HHT3 on chromosome 5 [9]. Of these, both ALK-1 and endoglin mutations are associated with an increased risk of PAH [7], and endoglin and, to a lesser extent, ALK-1 gene mutations lead to an increased risk of PAVMs. In HHT, the presence of both PAVMs and PAH is hence possible, with PAH characterised by an increase in Ppa leading to right heart failure and death [7, 10–12].
PAVMs may play a protective role in PAH. Like other potential right-to-left shunts, such as atrial septal defects, they provide a low-resistance pathway for blood flow and, therefore, could protect the right ventricle from increased afterload [13, 14]. Embolisation of PAVMs in patients with PAH can, therefore, theoretically lead to an increase in Ppa and right ventricular dysfunction. The risk of worsening pre-existing pulmonary hypertension following surgical PAVM closure was considered almost 40 yrs ago [12]. More recently, Haitjema et al. [15] have reported a case of increased mean Ppa confirmed by right heart catheterisation in a patient with PAH associated with HHT. However, the increase in mean Ppa in this patient was due, in part, to a high cardiac output secondary to a hepatic arteriovenous malformation, rather than a pulmonary vascular disease, and partial occlusion of the hepatic arteriovenous malformations led to a reduction in mean Ppa from 31 to 23 mmHg [15]. In a recent study, Shovlin et al. [16] have shown that embolisations of PAVMs did not lead to a consistent increase in mean Ppa in HHT patients without pulmonary hypertension, nor even in the setting of mild to moderate pre-existing pulmonary hypertension. However, patients with severe PAH (>35 mmHg) were not considered for embolisation in this series [16].
Since the routine use of radiologically inserted coils [17], very few studies have reported on the real risk of precipitating worse pulmonary hypertension, although PAVM embolisation is generally considered to be contraindicated in patients with severe PAH [4, 18]. Of a series of 66 patients who had a mean Ppa 15.3 mmHg (range 8–36 mmHg; mean systolic Ppa 26 mmHg and diastolic Ppa 8.8 mmHg) just prior to coil embolisation, the procedure only lead to an elevation in Ppa in one patient in whom the pulmonary hypertension was secondary to left ventricular disease [5]. A limitation of current studies on embolisation is the lack of haemodynamic data other than Ppa measurements, with no cardiac output and hence pulmonary vascular resistance (PVR) data. It has been shown that embolisation itself may reduce cardiac output [2] and this may, therefore, directly reduce Ppa. Some patients could experience an increase in PVR after embolisation without an observed increase in Ppa. Further studies with complete haemodynamic evaluation in HHT patients are needed to evaluate the risk of elevation of PVR.
In HHT, PAVMs tend to increase in size, especially if multiple PAVMs are present, and rarely regress spontaneously. They also tend to increase in size during pregnancy with the normal increase in cardiac output, blood volume and reduced vascular resistance [2]. Their increasing size is governed by Laplace's law, which describes the parietal wall tension (T) being dependent on the pressure (P) and diameter (2r) of the vessel, and inversely dependent on the thickness of the vessel wall (h), as defined by the following equation (fig. 4⇓):

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Laplace's law: T = (P×r)/2h. Pulmonary arteriovenous malformations (PAVMs) tend to increase in size because of the elevated parietal wall tension in the malformation. The increasing size of PAVM is governed by Laplace's law, which describes the parietal wall tension (T) being dependent on the pressure (P) and diameter (2r) of the vessel, and inversely dependent on the thickness of the vessel wall (h). In pulmonary arterial hypertension, the increase of pulmonary pressure leads to an increase in parietal wall tension that could lead to an excess risk of PAVM rupture.
T = (P×r)/2h (1)
In PAH, the increase in Ppa leads to an increase in parietal wall tension in the malformation, with afferent arteriolar dilatation and, ultimately, to a thin-walled aneurysmal sac susceptible to rupture. PAH is generally considered as a pro-thrombotic disease with microthrombotic pulmonary vascular lesions, and anticoagulation is recommended [19]. In patients with both PAH and PAVMs, this may present a management challenge, balancing an increased risk of PAH disease progression and also embolic events relating to the PAVM against the risk of massive haemoptysis or haemothorax.
Severe pulmonary arterial hypertension is generally considered a contraindication to performing embolisation of pulmonary arteriovenous malformations because of the risk of worsening of pulmonary arterial hypertension and precipitating right heart failure. However, given the excess risk of rupture, paradoxical embolism and haemoptysis, and the lack of data regarding the evolution of pulmonary artery pressure after embolisation in pulmonary arterial hypertension, pulmonary arteriovenous malformation embolisation might be considered in patients with stable pulmonary arterial hypertension. The balloon occlusion test, performed just prior to embolisation, gives a pre-procedure evaluation of haemodynamics that could help to discriminate which patients would develop an increase in pulmonary pressure following embolisation. A similar test is used in patients undergoing atrial septal defect repair [20], as well as in other endovascular procedures; however, there are few data on its use in pulmonary arteriovenous malformation embolisation [18]. This is a potentially useful test for predicting post-procedure pulmonary hypertension, but further validation is required, as well as the need for further studies to evaluate the real risk of embolising pulmonary arteriovenous malformations in patients with severe pulmonary arterial hypertension.
Support statement
L.C. Price is supported by a long-term research fellowship from the European Respiratory Society (number 139).
Statement of interest
None declared.
Provenance
Submitted article, peer reviewed.
Acknowledgments
The authors thank F. Soubrier (Laboratoire d'Oncogénétique et Angiogénétique Moléculaire, Groupe Hospitalier Pitié-Salpêtrière; Université Pierre et Marie Curie-Paris 6, Paris, France) for activin receptor-like kinase-1 gene mutation analysis.
- Received March 17, 2008.
- Accepted November 11, 2008.
- © ERSJ Ltd
References









